Development and application of adaptive, dual-resolution simulation methods
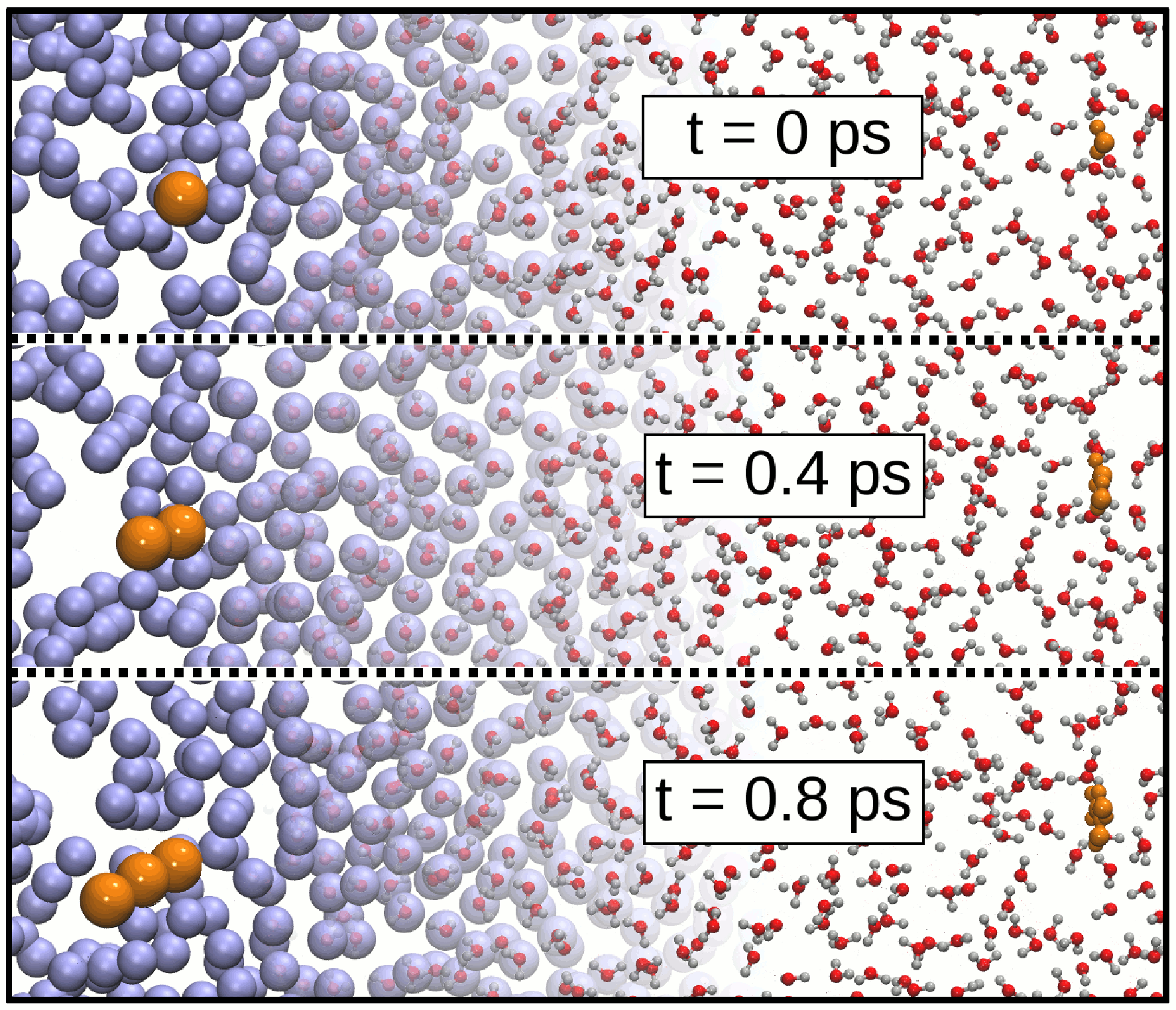
Many phenomena occurring in soft matter cover a broad range of length and time scales; to study them in simulations, a fully-atomistic description is computationally too expensive, yet coarse-grained models in many cases lack important chemical details. To circumvent this problem, adaptive resolution simulation methods have been developed to concurrently use different levels of resolution in the same simulation. Our group has largely contributed to the development of the Hamiltonian Adaptive Resolution Simulation (H-AdResS) scheme, which is based on a Hamiltonian function; this allows one to use the preferred statistical ensemble as well as the simulation algorithm, and makes it possible to formulate a solid statistical physics theory of dual-resolution systems. We employ this method to perform simulations of liquid systems and characterize their physical and thermodynamical properties.
Topological entanglement in biopolymers
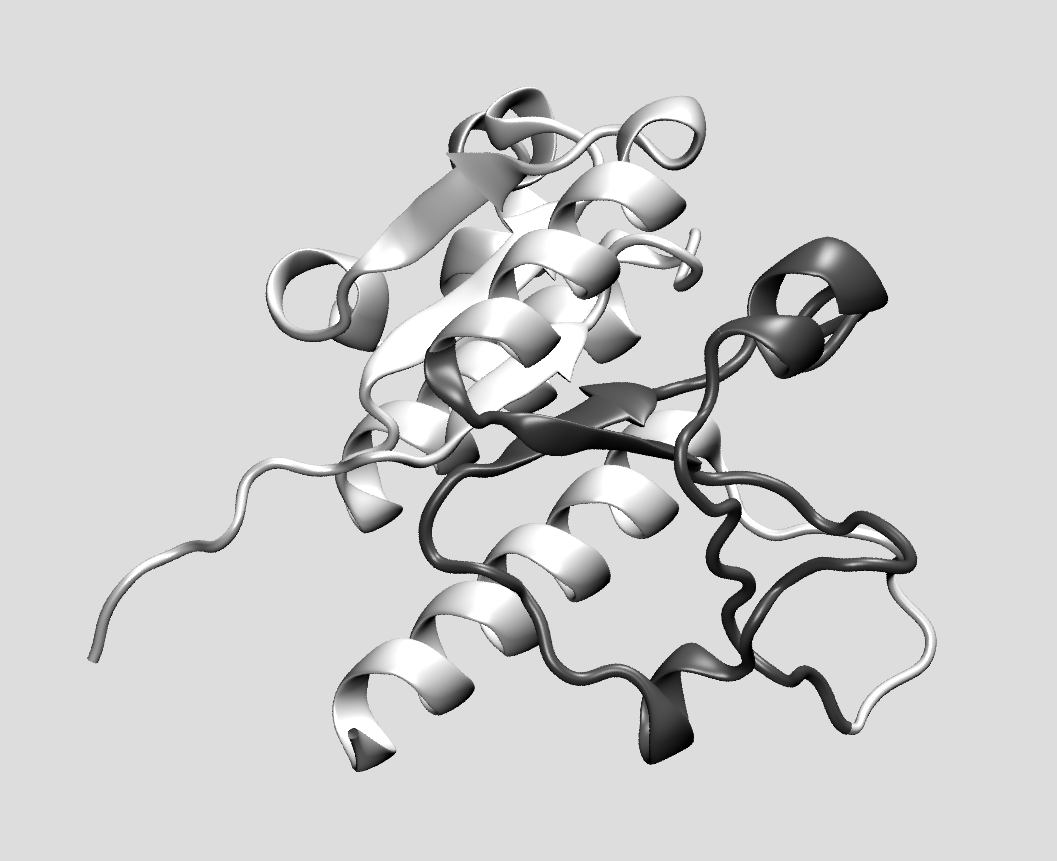
If protein folding represents on its own right a difficult scientific puzzle, the fact that some proteins (more than a hundred) possess a knotted native state is possibly even more puzzling. The sequence of events that take an unfolded polypeptide to a specific, topologically complex state is in most cases unclear, and a numerical investigation is made even more difficult than usual by the longer time scales involved and the presence of topological kinetic traps. Making use of coarse-grained models of a biopolymer we try to understand how to let the chain fold in a selected knotted state and which pathway (or pathways) can lead to it. We also focus on the conformational properties of self-entangled polymers and the impact that single or multiple knots have on chains in equilibrium as well as under external driving.
Dual-resolution simulation of biomolecules
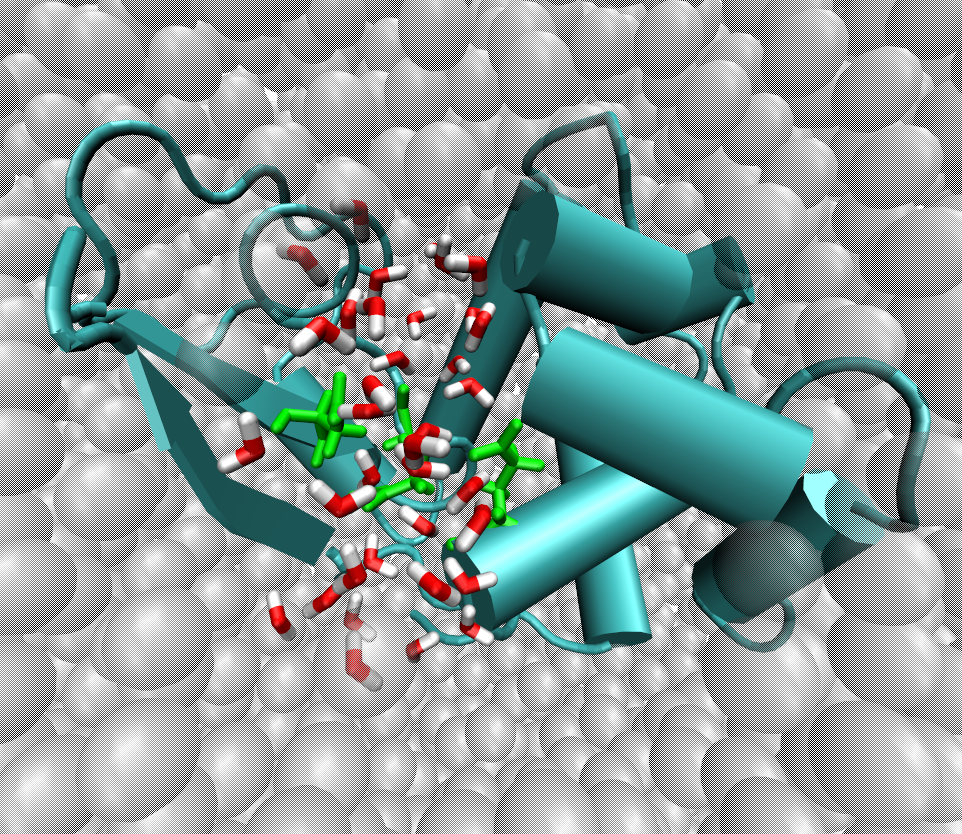
A prototypical example of a multiscale system is provided by large biomolecules, e.g. proteins, where the interplay of time and length scales makes it difficult, if not impossible, to perform atomistic simulations of a duration sufficient to observe physically relevant phenomena. At the same time, coarse-grained models might prove insufficient to model fine details whose accurate description is indispensable to understand the relation between a protein's chemical activity and its large-scale dynamics. An accurate description of an enzyme's active site can be joined by a coarse model of the parts of the protein whose chemical detail is not relevant, thus reducing the computational demand to simulate the system: dual-resolution simulation methods are currently being employed to achieve this goal.
This project is carried out in collaboration with Kurt Kremer and Aoife Fogarty within the framework of the ERC grant awarded to Kurt Kremer.