The calculation of free energy differences is a crucial step in the characterization and understanding of the physical properties of biological molecules. In the development of efficient methods to compute these quantities, a promising strategy is that of employing a dual-resolution representation of the solvent, specifically using an accurate model in the proximity of a molecule of interest and a simplified description elsewhere. One such concurrent multi-resolution simulation method is the Adaptive Resolution Scheme (AdResS), in which particles smoothly change their resolution on-the-fly as they move between different subregions. Before using this approach in the context of free energy calculations, however, it is necessary to make sure that the dual-resolution treatment of the solvent does not cause undesired effects on the computed quantities. Here, we show how AdResS can be used to calculate solvation free energies of small polar solutes using Thermodynamic Integration (TI). We discuss how the potential-energy-based TI approach combines with the force-based AdResS methodology, in which no global Hamiltonian is defined. The AdResS free energy values agree with those calculated from fully atomistic simulations to within a fraction of kBT. This is true even for small atomistic regions whose size is on the order of the correlation length, or when the properties of the coarse-grained region are extremely different from those of the atomistic region. These accurate free energy calculations are possible because AdResS allows the sampling of solvation shell configurations which are equivalent to those of fully atomistic simulations. The results of the present work thus demonstrate the viability of the use of adaptive resolution simulation methods to perform free energy calculations and pave the way for large-scale applications where a substantial computational gain can be attained.
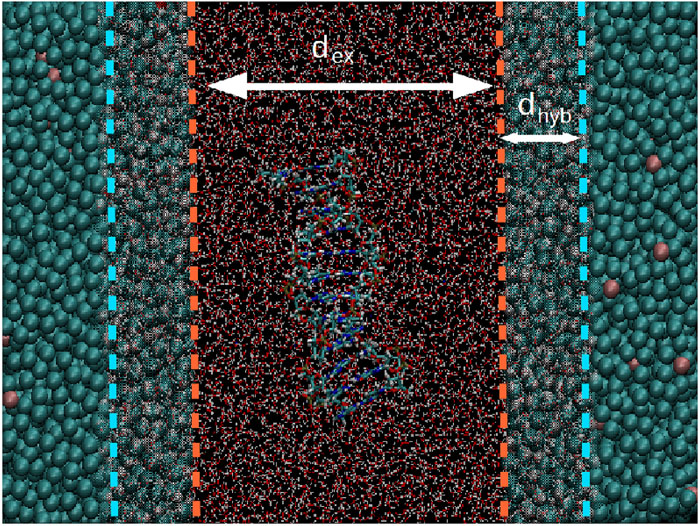
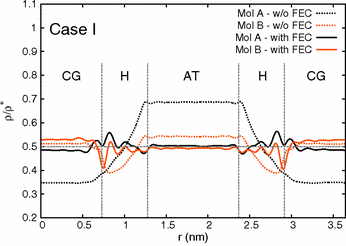
Nucleic acids are characterized by a complex hierarchical structure and a variety of interaction mechanisms with other molecules. These features suggest the need of multiscale simulation methods in order to grasp the relevant physical properties of deoxyribonucleic acid (DNA) and RNA using in silico experiments. Here we report an implementation of a dual-resolution modeling of a DNA oligonucleotide in physiological conditions; in the presented setup only the nucleotide molecule and the solvent and ions in its proximity are described at the atomistic level; in contrast, the water molecules and ions far from the DNA are represented as computationally less expensive coarse-grained particles. Through the analysis of several structural and dynamical parameters, we show that this setup reliably reproduces the physical properties of the DNA molecule as observed in reference atomistic simulations. These results represent a first step towards a realistic multiscale modeling of nucleic acids and provide a quantitatively solid ground for their simulation using dual-resolution methods.
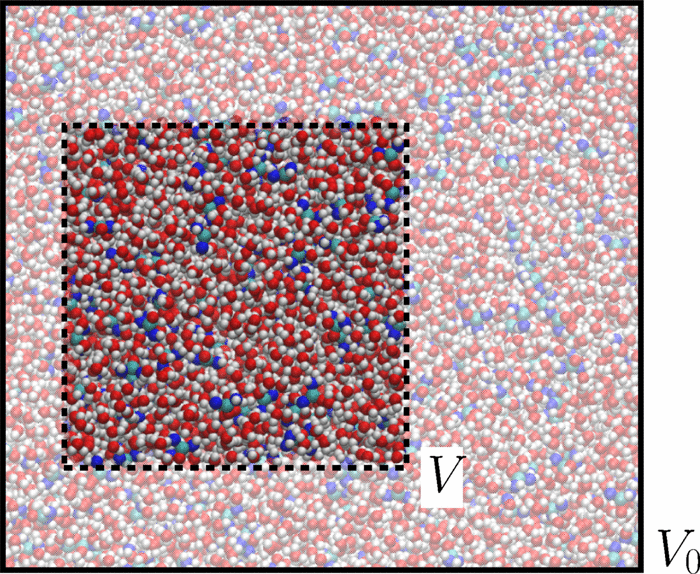
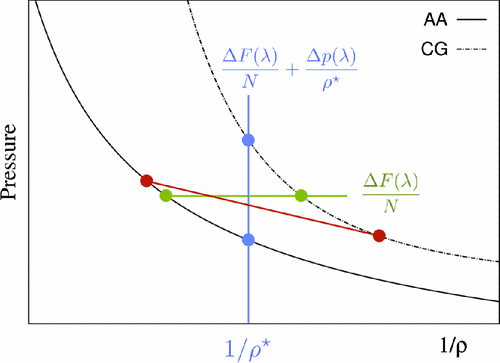
We present an accurate and efficient method to obtain Kirkwood-Buff (KB) integrals in the thermodynamic limit from small-sized molecular dynamics simulations. By introducing finite size effects into integral equations of statistical mechanics, we derive an analytical expression connecting the KB integrals of the bulk system with the fluctuations of the number of molecules in the corresponding closed system. We validate the method by calculating the activity coefficients of aqueous urea mixtures and the KB integrals of Lennard-Jones fluids. Moreover, our results demonstrate how to identify simulation conditions under which computer simulations reach the thermodynamic limit.
In multi-resolution simulations, different system components are simultaneously modeled at different levels of resolution, these being smoothly coupled together. In the case of enzyme systems, computationally expensive atomistic detail is needed in the active site to capture the chemistry of ligand binding. Global properties of the rest of the protein also play an essential role, determining the structure and fluctuations of the binding site; however, these can be modeled on a coarser level. Similarly, in the most computationally efficient scheme only the solvent hydrating the active site requires atomistic detail. We present a methodology to couple atomistic and coarse-grained protein models, while solvating the atomistic part of the protein in atomistic water. This allows a free choice of which protein and solvent degrees of freedom to include atomistically. This multi-resolution methodology can successfully model stable ligand binding, and we further confirm its validity by exploring the reproduction of system properties relevant to enzymatic function. In addition to a computational speedup, such an approach can allow the identification of the essential degrees of freedom playing a role in a given process, potentially yielding new insights into biomolecular function.
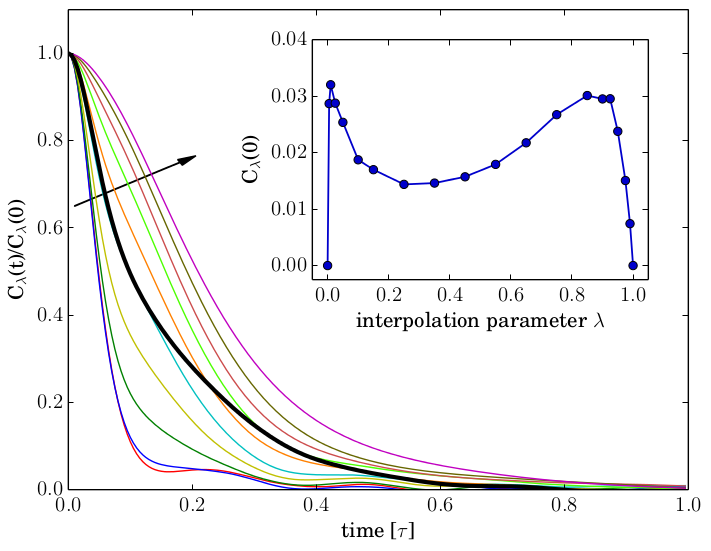
Adaptive resolution techniques are powerful methods for the efficient simulation of soft matter systems in which they simultaneously employ atomistic and coarse-grained (CG) force fields. In such simulations, two regions with different resolutions are coupled with each other via a hybrid transition region, and particles change their description on the fly when crossing this boundary. Here we show that the relative entropy, which provides a fundamental basis for many approaches in systematic coarse-graining, is also an effective instrument for the understanding of adaptive resolution simulation methodologies. We demonstrate that the use of coarse-grained potentials which minimize the relative entropy with respect to the atomistic system can help achieve a smoother transition between the different regions within the adaptive setup. Furthermore, we derive a quantitative relation between the width of the hybrid region and the seamlessness of the coupling. Our results do not only shed light on the what and how of adaptive resolution techniques but will also help setting up such simulations in an optimal manner.
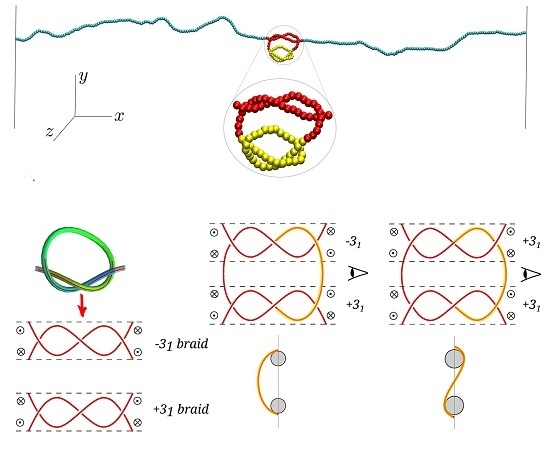
Knots appear frequently in semiflexible (bio)polymers, including double-stranded DNA, and their presence can affect the polymer's physical and functional properties. In particular, it is possible and indeed often the case that multiple knots appear on a single chain, with effects which have only come under scrutiny in the last few years. In this manuscript, we study the interaction of two knots on a stretched semiflexible polymer, expanding some recent results on the topic. Specifically, we consider an idealization of a typical optical tweezers experiment and show how the bending rigidity of the chain-And consequently its persistence length-Influences the distribution of the entanglements; possibly more importantly, we observe and report how the relative chirality of the otherwise identical knots substantially modifies their interaction. We analyze the free energy of the chain and extract the effective interactions between embedded knots, rationalizing some of their pertinent features by means of simple effective models. We believe the salient aspect of the knot-knot interactions emerging from our study will be present in a large number of semiflexible polymers under tension, with important consequences for the characterization and manipulation of these systems-Be they artificial or biologica in origin-And for their technological application.
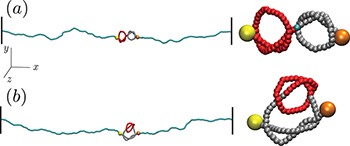
In this study we consider an idealization of a typical optical tweezers experiment involving a semiflexible double-knotted polymer, with steric hindrance and persistence length matching those of dsDNA in high salt concentration, under strong stretching. Using exhaustive molecular-dynamics simulations we show that not only does a double-knotted dsDNA filament under tension possess a free-energy minimum when the two knots are intertwined, but also that the depth of this minimum depends on the relative chirality of the two knots. We rationalize this dependence of the effective interaction on the chirality in terms of a competition between chain entropy and bending energy.
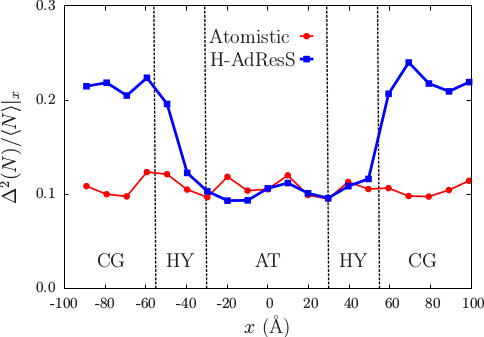
In adaptive resolution simulations the same system is concurrently modeled with different resolution in different subdomains of the simulation box, thereby enabling an accurate description in a small but relevant region, while the rest is treated with a computationally parsimonious model. In this framework, electrostatic interaction, whose accurate treatment is a crucial aspect in the realistic modeling of soft matter and biological systems, represents a particularly acute problem due to the intrinsic long-range nature of Coulomb potential. In the present work we propose and validate the usage of a short-range modification of Coulomb potential, the Damped shifted force (DSF) model, in the context of the Hamiltonian adaptive resolution simulation (H-AdResS) scheme. This approach, which is here validated on bulk water, ensures a reliable reproduction of the structural and dynamical properties of the liquid, and enables a seamless embedding in the H-AdResS framework. The resulting dual-resolution setup is implemented in the LAMMPS simulation package, and its customized version employed in the present work is made publicly available.

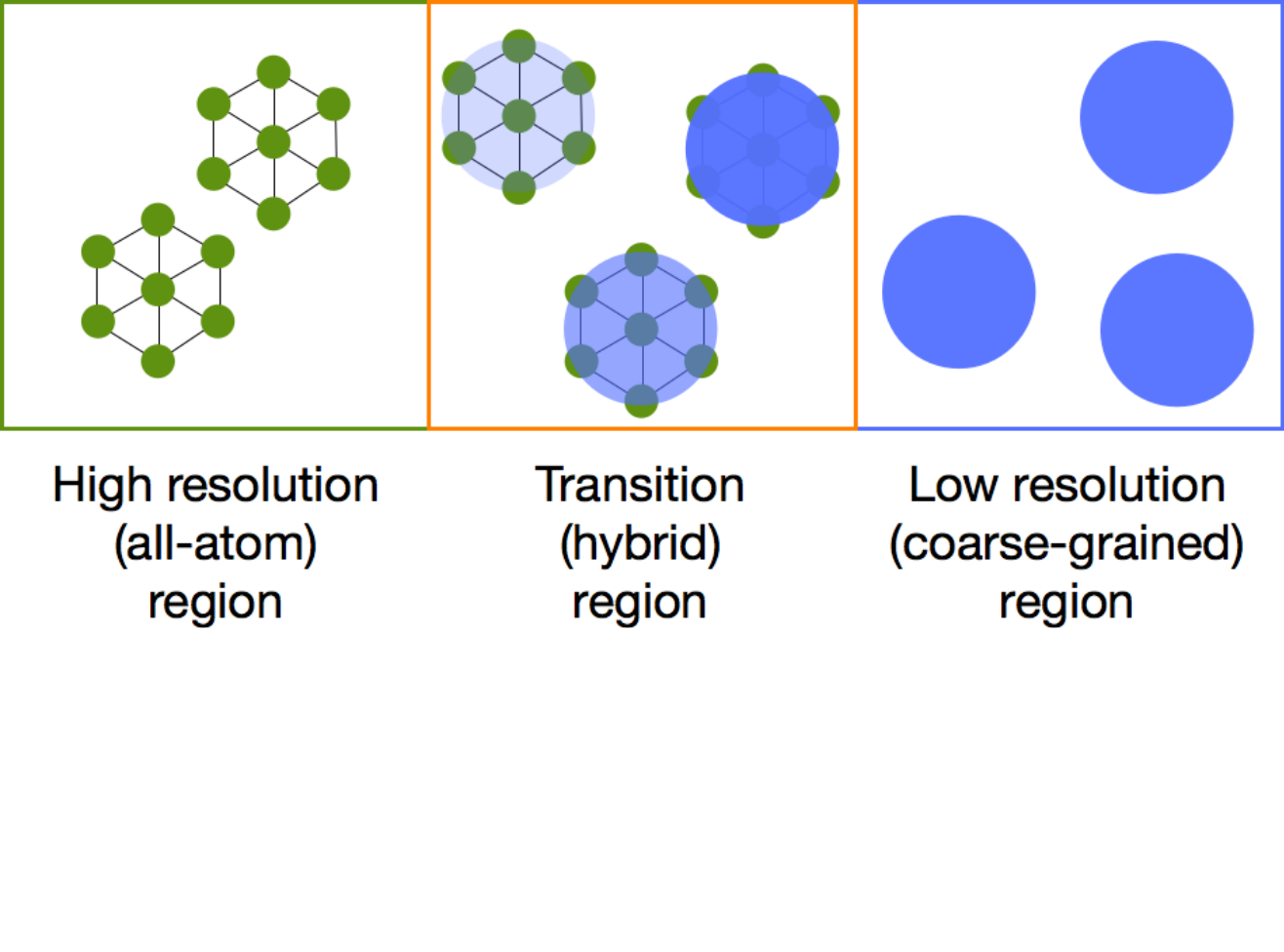
In adaptive resolution simulations, different regions of a simulation box are modeled with different levels of detail. Particles change their resolution on-the-fly when traveling from one subregion to the other. This method is particularly useful for studying multiscale systems in which effects on a broad range of length and time scales play a role. Until now, the geometry of the high-resolution region has been limited to simple geometries of spherical, cuboid, or cylindrical form, whose shape does not change during the simulation. However, many phenomena involve changes in size and shape of system components, for example, protein folding, polymer collapse, nucleation, and crystallization. In this work, we develop a scheme that uses a series of overlapping spheres to allow for an arbitrary division of space into domains of different levels of resolution. Furthermore, the geometry is automatically adjusted on-the-fly during the simulation according to changes in size and shape of, for example, a solvated macromolecule within the high-resolution region. The proposed approach is validated on liquid water. We then simulate the folding of an atomistically detailed polypeptide solvated in a shell of atomistic water that changes shape as the peptide conformation changes. We demonstrate that the peptide folding process is unperturbed by the use of our methodology.
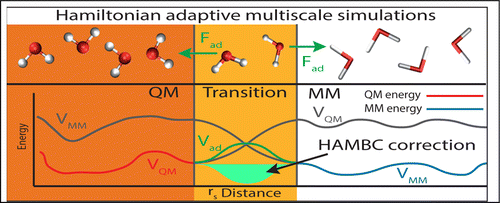
Adaptive quantum mechanical (QM)/molecular mechanical (MM) methods enable efficient molecular simulations of chemistry in solution. Reactive subregions are modeled with an accurate QM potential energy expression while the rest of the system is described in a more approximate manner (MM). As solvent molecules diffuse in and out of the reactive region, they are gradually included into (and excluded from) the QM expression. It would be desirable to model such a system with a single adaptive Hamiltonian, but thus far this has resulted in distorted structures at the boundary between the two regions. Solving this long outstanding problem will allow microcanonical adaptive QM/MM simulations that can be used to obtain vibrational spectra and dynamical properties. The difficulty lies in the complex QM potential energy expression, with a many-body expansion that contains higher order terms. Here, we outline a Hamiltonian adaptive multiscale scheme within the framework of many-body potentials. The adaptive expressions are entirely general, and complementary to all standard (nonadaptive) QM/MM embedding schemes available. We demonstrate the merit of our approach on a molecular system defined by two different MM potentials (MM/MM'). For the long-range interactions a numerical scheme is used (particle mesh Ewald), which yields energy expressions that are many-body in nature. Our Hamiltonian approach is the first to provide both energy conservation and the correct solvent structure everywhere in this system.
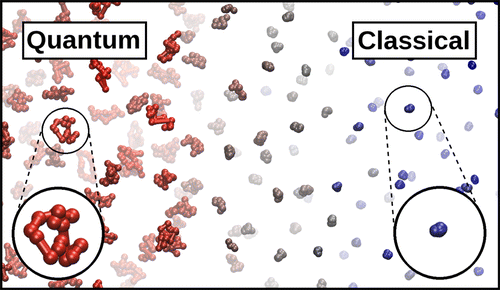
Quantum delocalization of atomic nuclei affects the physical properties of many hydrogen-rich liquids and biological systems even at room temperature. In computer simulations, quantum nuclei can be modeled via the path-integral formulation of quantum statistical mechanics, which implies a substantial increase in computational overhead. By restricting the quantum description to a small spatial region, this cost can be significantly reduced. Herein, we derive a bottom-up, rigorous, Hamiltonian-based scheme that allows molecules to change from quantum to classical and vice versa on the fly as they diffuse through the system, both reducing overhead and making quantum grand-canonical simulations possible. The method is validated via simulations of low-temperature parahydrogen. Our adaptive resolution approach paves the way to efficient quantum simulations of biomolecules, membranes, and interfaces.
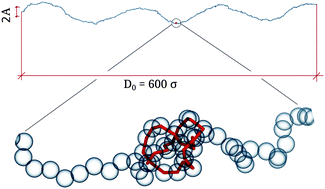
Topological entanglement is a ubiquitous feature of many biological as well as artificial polymers and fibers. While the equilibrium properties of entangled chains have been the subject of several studies, little is known about their out-of-equilibrium behavior. Here, we address the problem of a stretched knotted fiber driven by a periodic force applied to one of its termini. We show that the onset of standing waves kinetically traps the knot in spatially localized states where the amplitude of the oscillations is maximal, while the knot normal diffusive dynamics is replaced by a discrete jump dynamics.
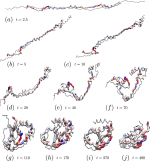
A small but relevant number of proteins whose native structure is known features nontrivial topology, i.e., they are knotted. Understanding the process of folding from a swollen unknotted state to the biologically relevant native conformation is, for these proteins, particularly difficult, due to their rate-limiting topological entanglement. To shed some light into this conundrum, we introduced a structure-based coarse-grained model of the protein, where the information about the folded conformation is encoded in bonded angular interactions only, which do not favor the formation of native contacts. A stochastic search scheme in parameter space is employed to identify a set of interactions that maximizes the probability to attain the knotted state. The optimal knotting pathways of the two smallest knotted proteins, obtained through this approach, are consistent with the results derived by means of coarse-grained as well as full atomistic simulations.
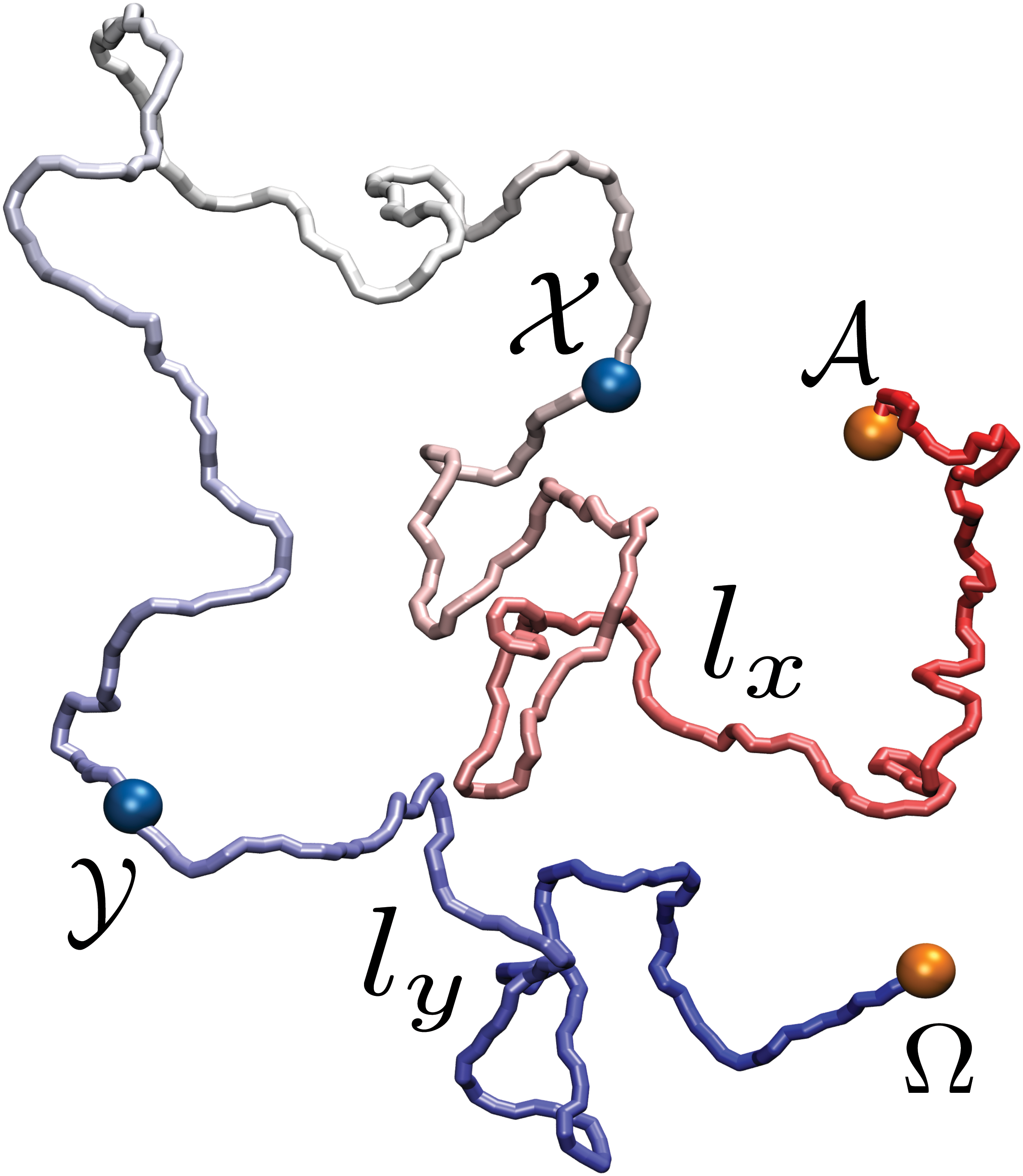
Self-entanglement, or knotting, is entropically favored in long polymers. Relatively short polymers such as proteins can knot as well, but in this case the entanglement is mainly driven by fine-tuned, sequence-specific interactions. The relation between the sequence of a long polymer and its topological state is here investigated by means of a coarse-grained model of DNA. We demonstrate that the introduction of two adhesive regions along the sequence of a self-avoiding chain substantially increases the probability of forming a knot.
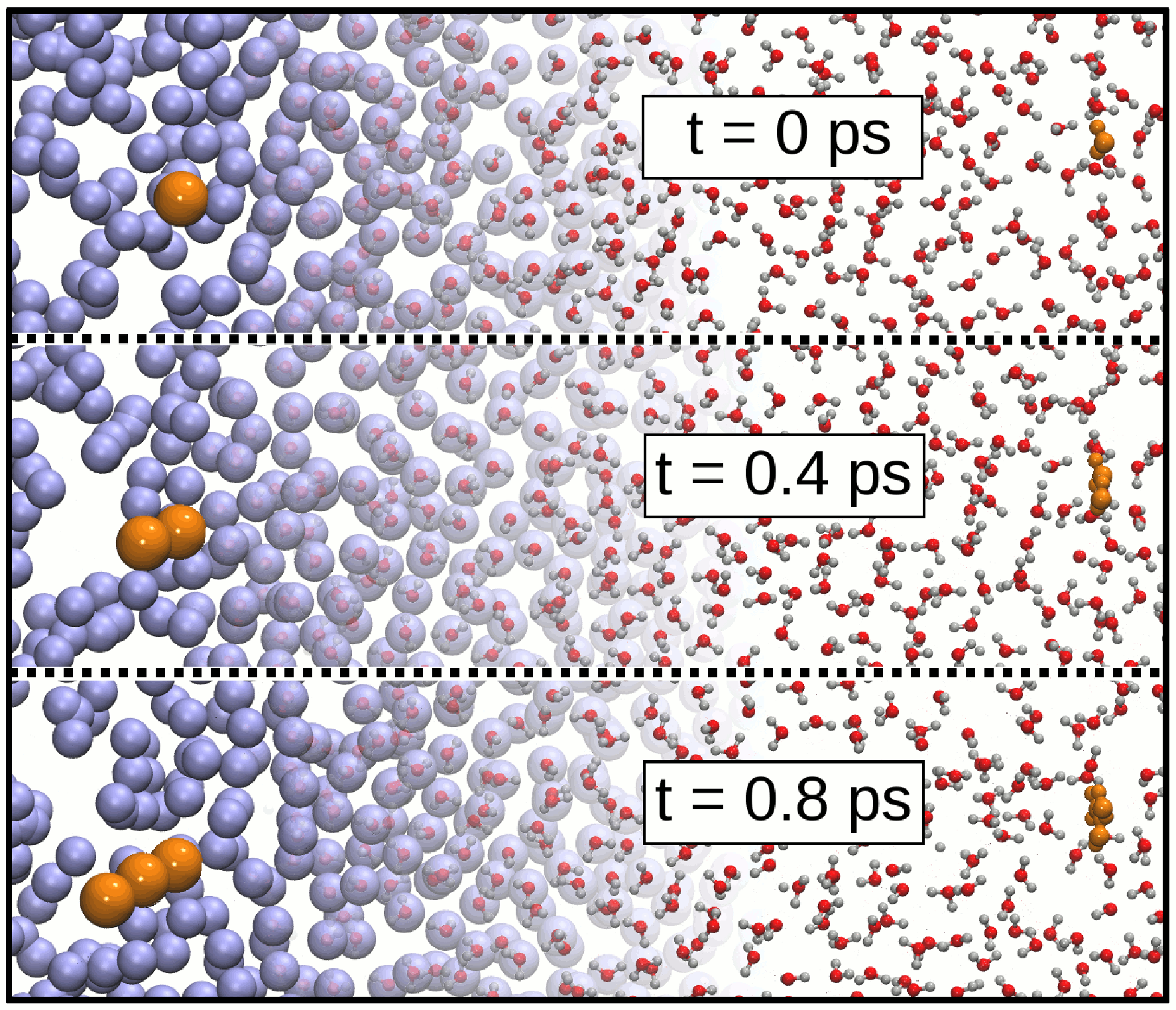
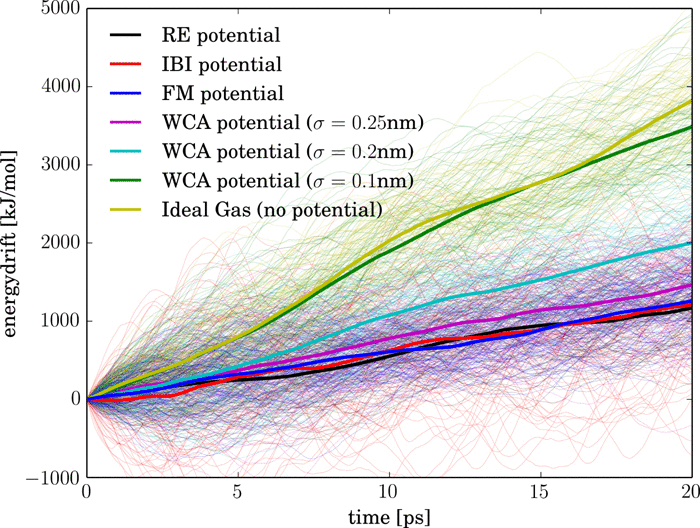
One of the main advantages of adaptive resolution simulations is the computational efficiency gained in the coarse-grained region. In this respect the best coarse-grained system would be the ideal gas, making intermolecular force calculations redundant. In this case, however, a smooth coupling is challenging due to the high energetic imbalance between typical liquids and a system of non-interacting particles. In the present work, we investigate this approach, using as a test case the most biologically relevant fluid, water. We demonstrate that a successful coupling of water to the ideal gas can be achieved with current adaptive resolution methods, and discuss the issues that remain to be addressed.
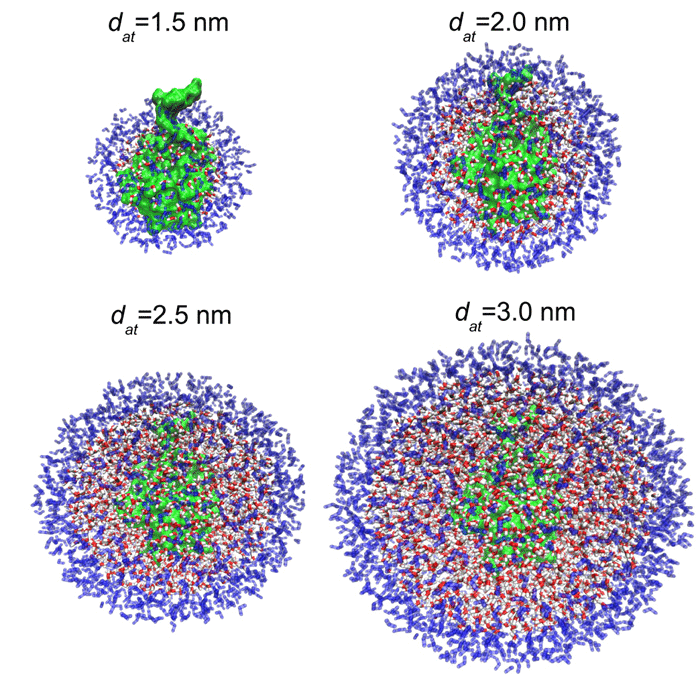
A fully atomistic modelling of many biophysical processes is often beyond our reach, and one approach to overcome this difficulty is the use of multiscale simulation techniques. Here, we apply the AdResS methodology to biomolecular systems, simulating a fully atomistic protein solvated in a coarse-grained particle reservoir and heat bath. Using as a test case an aqueous solution of ubiquitin, we first confirm the validity of the AdResS approach via an examination of structural and dynamical properties. We then demonstrate how such a multiscale approach can yield otherwise inaccessible physical insights into biomolecular function.
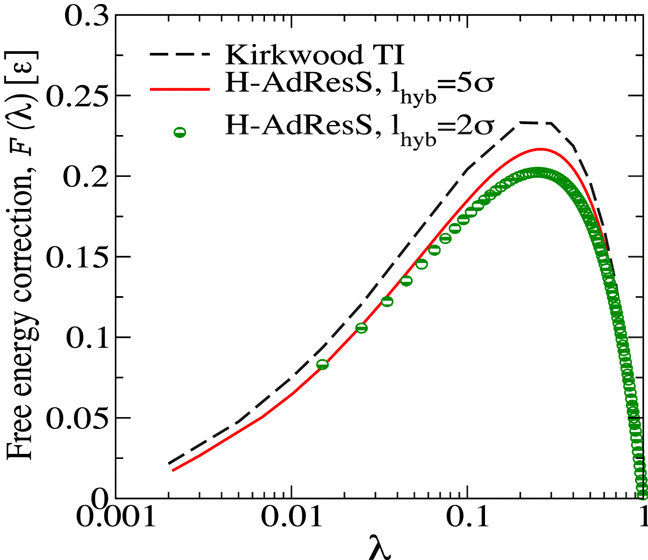
The Adaptive Resolution Scheme is a hybrid scheme that allows to treat a molecular system with different levels of resolution depending on the location of the molecules. The construction of a Hamiltonian based on the this idea (H-AdResS) allows one to formulate the usual tools of ensembles and statistical mechanics. We present a number of exact and approximate results that provide a statistical mechanics foundation for this simulation method. We also present simulation results that illustrate the theory.
Adaptive resolution simulation schemes are based on a position-dependent interpolation of either forces or potential energy functions. While force-based methods generally lead to non-conservative forces, energy-based ones include undesired force terms proportional to the gradient of the interpolation function. In this work we establish a so far missing bridge between these formalisms making use of the generalized Langevin equation, thereby providing a unifying framework to traditionally juxtaposed approaches to adaptive simulations.
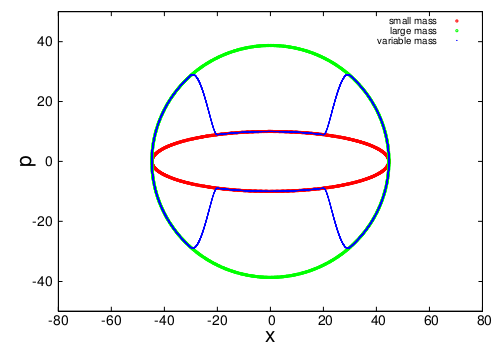
In the present work a dynamical system is investigated, in which the particles' mass depends on their position in space. A single point-like particle in one dimension and a Lennard-Jones fluid in three dimensions, whose Hamiltonian is numerically integrated with a first-order, energy-conserving algorithm, are studied. The features of both setups are examined, and a simple, exact method is devised to obtain the same equilibrium observables that would be measured in a conventional simulation.

In this review we discuss the basic notions of coarse-graining theory, presenting the most common methodologies employed to build low-resolution descriptions of a system and putting particular emphasis on their similarities and differences. The AdResS and H-AdResS adaptive resolution simulation schemes are reported as examples of dual-resolution approaches, especially focusing in particular on their theoretical background.
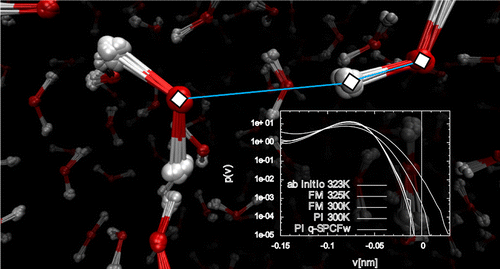
We outline a computationally efficient method to investigate the role of nuclear quantum effects in liquid water making use of a force field derived from ab initio simulations. The model, which reproduces the key structural and dynamic properties of the reference system, was used to perform path integral simulations to investigate the role played by nuclear quantum effects on water.
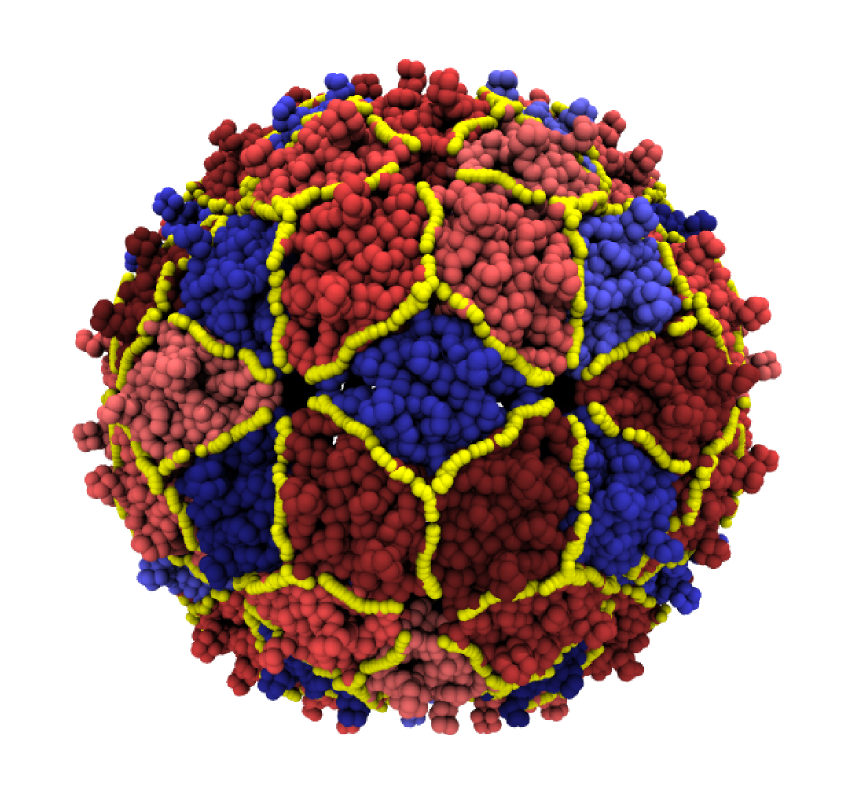
We introduce and apply a general and efficient computational scheme for identifying the stable domains of a given viral capsid. The method, based on elastic network models and quasi-rigid domain decomposition, is first applied to a heterogeneous set of well-characterized viruses for which the known mechanical or assembly domains are correctly identified; the validated scheme is next applied to other viral particles whose fundamental functional domains are still unknown and for which we formulate verifiable predictions.
Complex soft matter systems can be efficiently studied with the help of adaptive resolution simulation methods, concurrently employing two levels of resolution in different regions of the simulation domain. In this work we employ the Hamiltonian adaptive resolution simulation method to perform Monte Carlo simulations of a binary mixture, and propose an efficient scheme to regulate the thermodynamic balance of multicomponent systems.
Adaptive resolution schemes allow the simulation of a molecular fluid treating simultaneously different subregions of the system at different levels of resolution. Here we present a new scheme, formulated in terms of a global Hamiltonian, where equilibrium states corresponding to well-defined statistical ensembles can be generated making use of all standard molecular dynamics or Monte Carlo methods. Models at different resolutions can thus be coupled and thermodynamic equilibrium can be modulated.
This lecture provides an introduction to the Adaptive Resolution Simulation (AdResS) and the Hamiltonian AdResS (H-AdResS) schemes, which have been developed to simulate a subregion of the system with a higher resolution with respect to a surrounding thermal bath of coarser molecules; open boundaries between these regions guarantee that the correct thermodynamics is preserved in the high-resolution region of interest.
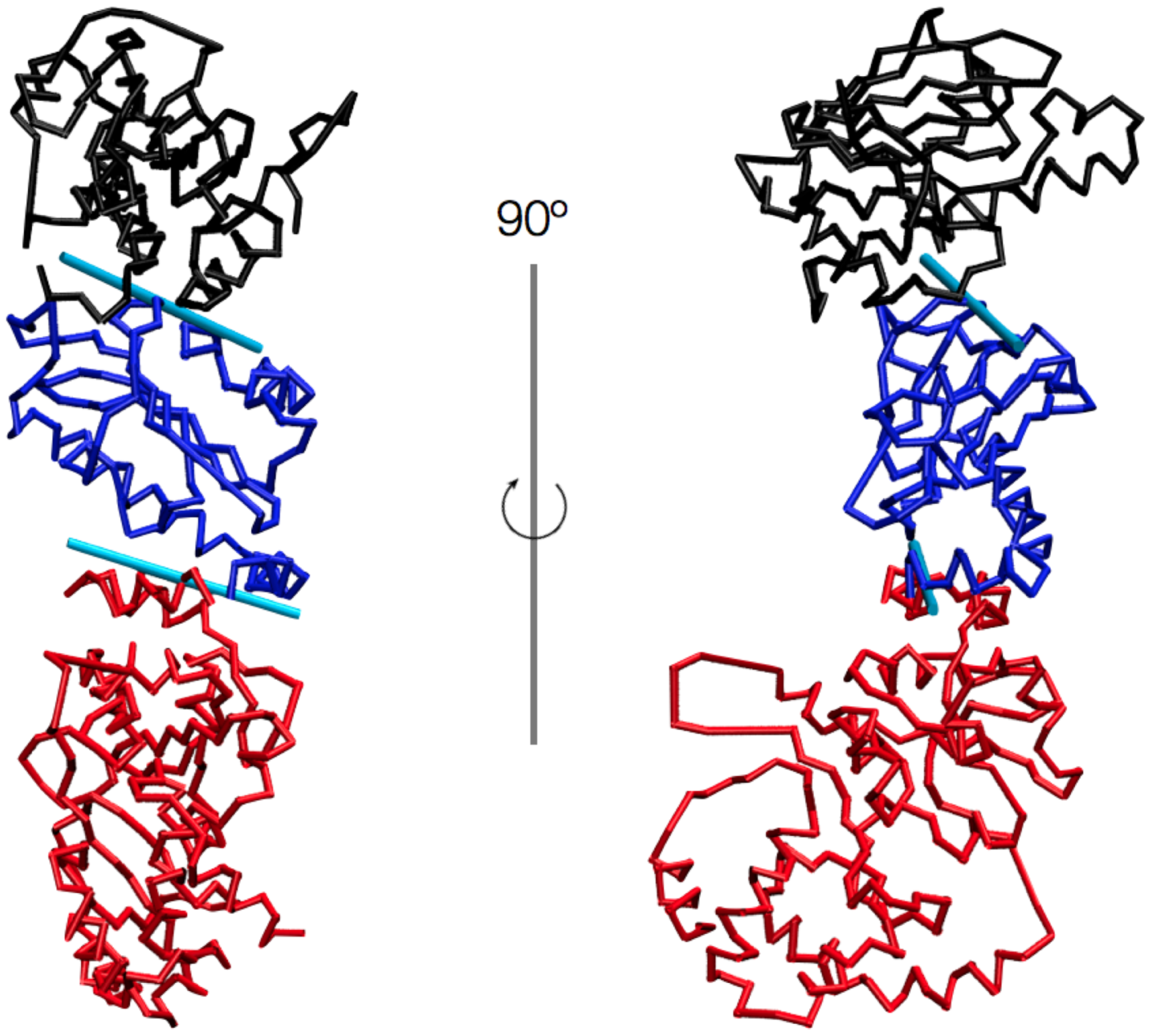
Understanding how local protein modifications can trigger and regulate large-scale motions of large protein domains is a major open issue in molecular biology. We address various aspects of this problem by analyzing and comparing atomistic simulations of three Hsp90 family representatives with a novel multi-scale comparison of their internal dynamics. The results reveal that the ligand-dependent structural modulations mostly consist of relative rigid-like movements of a limited number of domains shared by the three proteins.

Parahydrogen is the spin-zero singlet state of molecular hydrogen, which at low temperature (between 14 and 25 K) is in a fluid state. A classical treatment of the system leads to unphysical freezing, and the inclusion of quantum delocalization of the molecule is then required to obtain a realistic description of its equilibrium properties. In the present work, the classical-quantum adaptive resolution method AdResS is employed to investigate the spatial extension of quantum delocalization effects in the bulk fluid at low temperature.