|
|
| |
``............ everything that living things do can be
understood in terms of the jiggling and wiggling of atoms" - Richard Feynman
MULTI-RESPONSIVE SMART POLYMERS:
Polymer processing requires a detailed understanding of their structure-property relationships,
which not only is fundamentally challenging but also has a wide range of applications ranging from
physics to biology. Here, smart responsive polymers serve as excellent candidates.
A polymer is commonly known as ``smart responsive" when a small change in external stimulus can drastically
change its structure, function and/or stability.
However, for a given smart homopolymer structure, there exists a characteristic transition temperature.
This transition temperature can be tuned by introducing more hydrophobic or more hydrophilic units along
the homopolymer backbone. In particular, transition temperature increases (decreases) with increasing hydrophilic (hydrophobic) units.
Moreover, the changes in transition temperatures are usually difficult to predict and are non-linear with changing copolymer sequences.
Furthermore, polymer properties are inherently multi-scale in nature,
where delicate local interaction details play a key role in describing their global conformational behavior.
Further complexities arise when dealing with copolymer systems with varying microscopic
sequences, especially when they are of an amphiphilic nature.
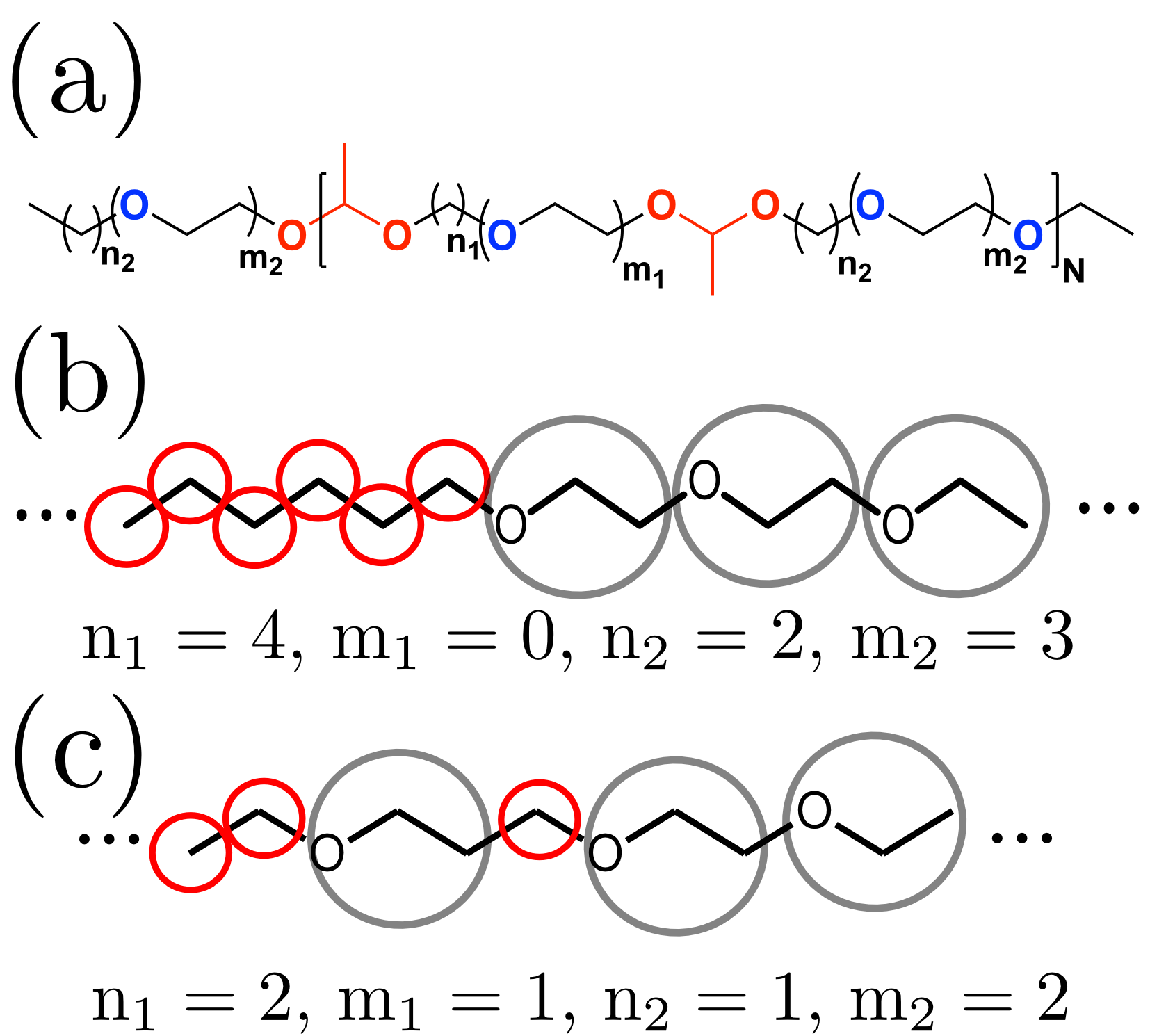
In this work we have so far developed a segment-based CG model for acetal based amphiphilic copolymers synthesized recently in
the Koberstein Group (Columbia University), as shown in part (a) of the figure.
This CG model (schemes shown in parts (b) and (c) of the figure) helps interpreting and guiding experiment towards new directions. This hand-in-hand work based on solid theoretical
grounds is quite rare and can help as a guiding principle for the design of a new class of multi-responsive polymers.
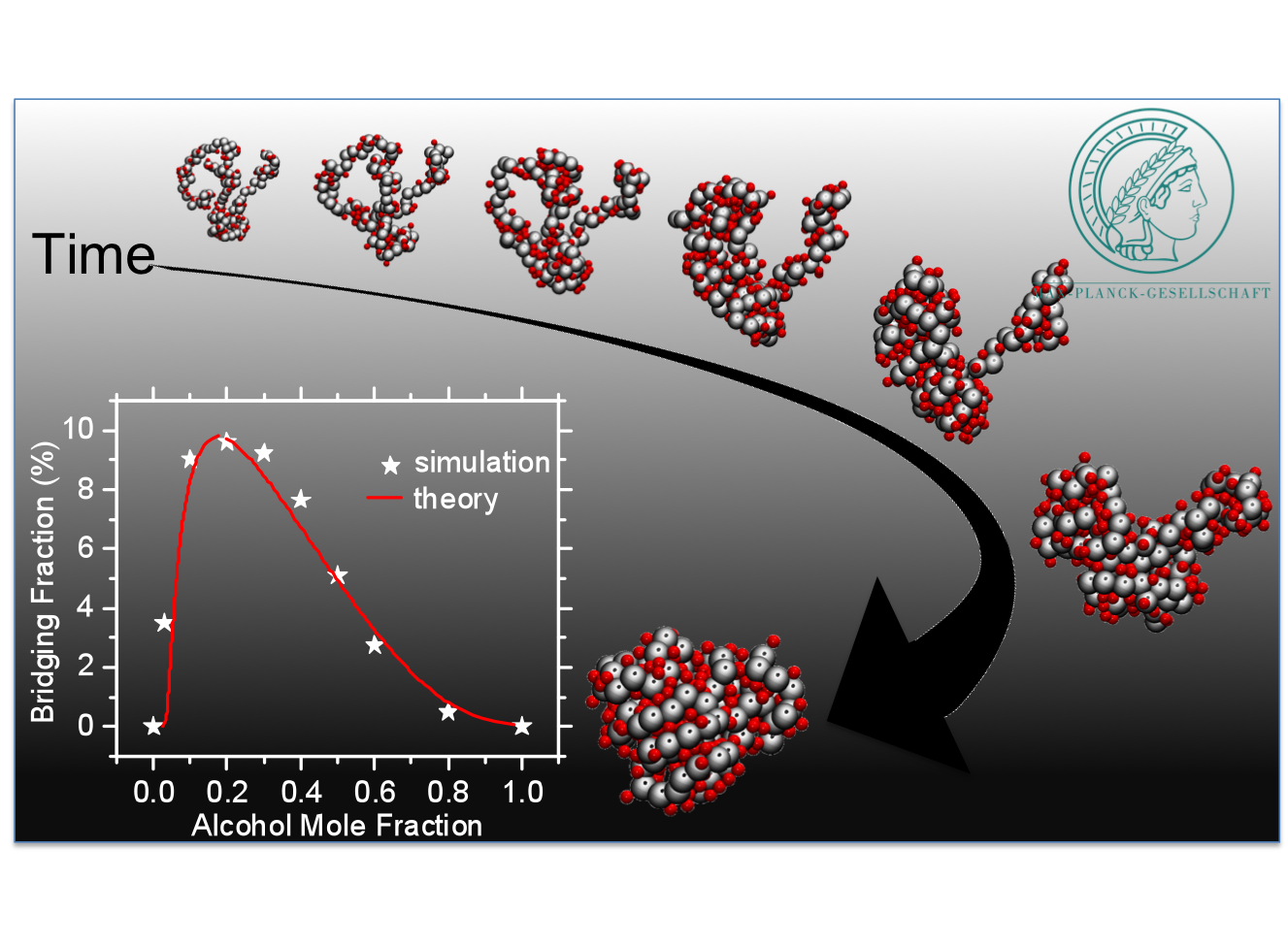
SMART POLYMERS IN MIXED SOLVENTS:
Stimuli responsive polymers, often referred to as smart polymers, are macromolecular systems whose structure,
function and stability are intimately linked to external parameters, such as temperature, external fields, ionic strengths
or cosolvent concentrations. This is the origin of the responsiveness of hydrogels exposed to external stimuli or
concentration gradients and denaturation of proteins. Most common stimuli responsive polymers display a transition
from an extended coil structure at low temperature to a compact globule when temperature is increased, thus presenting
a lower critical solution temperature (LCST). Another interesting aspect is when these polymers are solvated in
mixed (co)solvents. More specifically, water and alcohol are miscible and, individually, good solvents for
these smart polymers, but the same polymers precipitate in water-alcohol mixtures. The intriguing behavior of solvent
mixtures that cannot dissolve a given polymer or a given protein, while the same macromolecule dissolves well
in each of the cosolvents, is called cononsolvency. It is a widespread phenomenon, relevant for many
formulation steps in the physicochemical and pharmaceutical industry, that is usually explained by
invoking specific chemical details of the mixtures: as such it has so far eluded any generic
explanation. In this project, by using a combination of computer simulations and analytical theory,
we have proposed a simple and universal treatment that requires only the preferential interaction
of one of the cosolvents with the polymer. Because of this discrete particle based nature of the
interactions, Flory-Huggins type mean field arguments become unsuitable. Therefore, co-non-solvency
can only be understood by the thermodynamic treatment of the competitive displacement of (co)solvent components.
This competition can result in a polymer collapse upon improvement of the solvent quality. Specific chemical details
are not required to understand these complex conformational transitions. Therefore, a broad range of polymers
are expected to exhibit similar conformational transition in competing good solvents.
Our study opens a new perspective towards an operational understanding of macromolecular solubility.
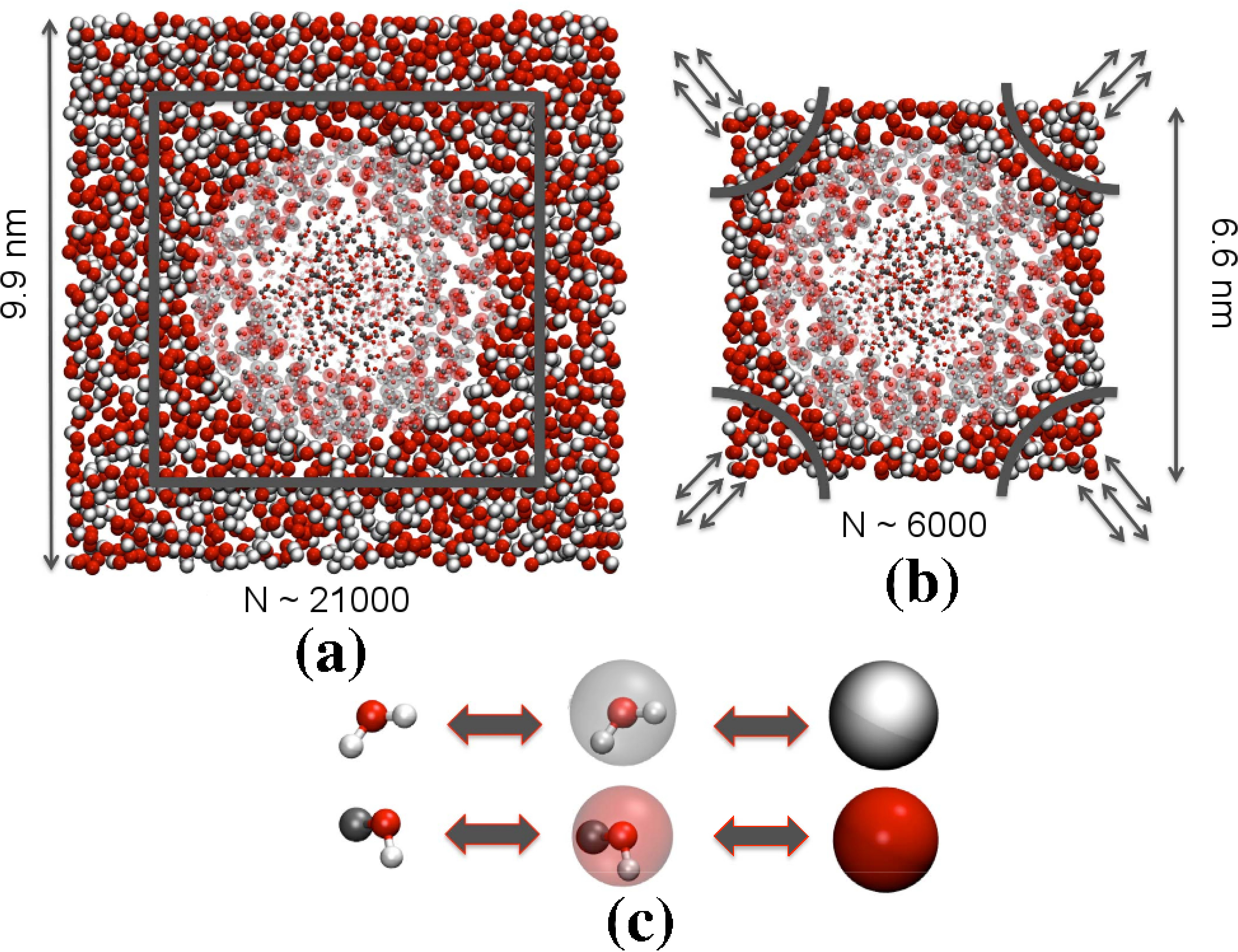
SEMI-GRAND CANONICAL MOLECULAR DYNAMICS:
Conformational transitions of (bio)macromolecules in aqueous mixtures are intimately linked to local
concentration fluctuations of different solvent components. Though computer simulations are ideally
suited to investigate such phenomena, in conventional setups the excess of one cosolvent close to the
solute leads to depletion elsewhere, disturbing the solvent equilibrium. However, the solvent equilibrium can
either be maintained by choosing an enormously large solvent bath or by employing a grand-canonical
type particle insertion protocol within a small-sized simulation domain.
Moreover, when dealing with all-atom representations of complex molecular solvents, the replacements
of molecules is hindered by poor acceptance rates. If, instead, the molecules are represented by
spherically symmetric CG beads, replacement of another bead is typically easier. Therefore,
we have developed a semi-grand canonical method for (bio)macromolecular simulations. Our
method makes use of the adaptive resolution scheme (AdResS) scheme in conjunction with the Metropolis particle exchange
criterion, which is performed within a CG region of the simulation setup. We have tested our method to study
the experimentally observed reentrant coil-globule-coil transition of poly(N-isopropylacrylamide) in
aqueous methanol mixtures.
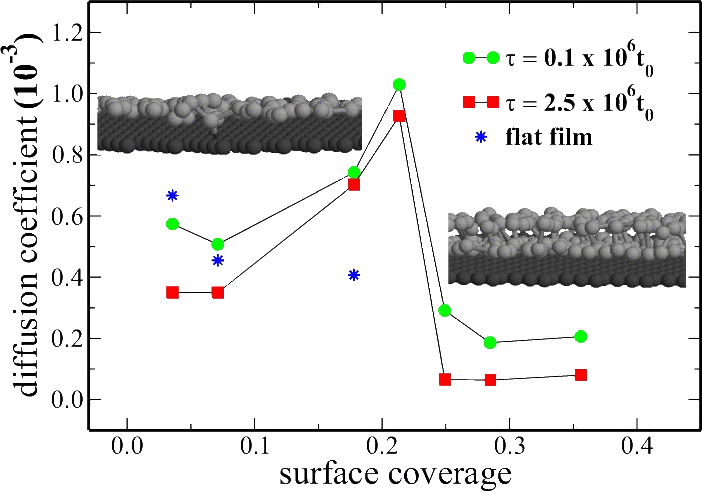
DIFFUSION AND AGING OF POLYMER FILMS NEAR SURFACES:
The image shows the lateral diffusion coefficient for strongly adsorbed mono-dispersed
polymer/ oligomer chains. Anomalous behavior is a result of structural transition
from single to double layer, which gives polymer more geometrical flexibility to adopt
to the surface corrugation and hence, within the Arrhenius type activation picture, is
responsible for much reduced lateral mobility at large concentrations. The graph also
contains information about the aging effects (non-equilibrium dynamics). A distinct
reduction in mobility is observed, at all surface coverages, for the system with
increasing ages.
SINGLE POLYMER DYNAMICS IN HIGH GEOMETRIC CONFINEMENTS:
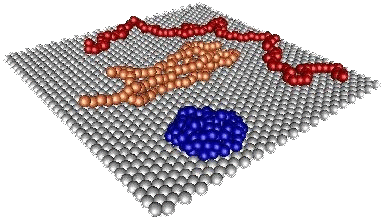
The dynamics of polymer chains near surfaces has attracted increased attention over last decade.
While early studies strived to achieve a fundamental understanding of the often exotic
and unpredictable behavior of polymers in confinement, recent research has mostly been
motivated by applications that range from biology to tribology. Despite significant progress
within the last decade, in particular on the structural and thermodynamic properties of
polymers in confinement, their dynamics have remained poorly understood. We used coarse-grained
molecular dynamics simulations to study various controversial and unexplained experiments
reported in the literature. We included variable atomic scale roughness of the confining
surfaces while abstaining from introducing any artificially-imposed, divergent obstacles.
Because of this type of modeling, we achieved striking qualitative agreement with
existing experimental results on the scaling of single-polymer mobility near surfaces
with the degree of polymerization. In the simulation snapshot, we show polymer in good
solvent (red), poor solvent (blue) and unstable knot (orange).
NEAT THERMOSET AND THERMOSET/THERMOPLASTIC POLYMER ALLOYS::
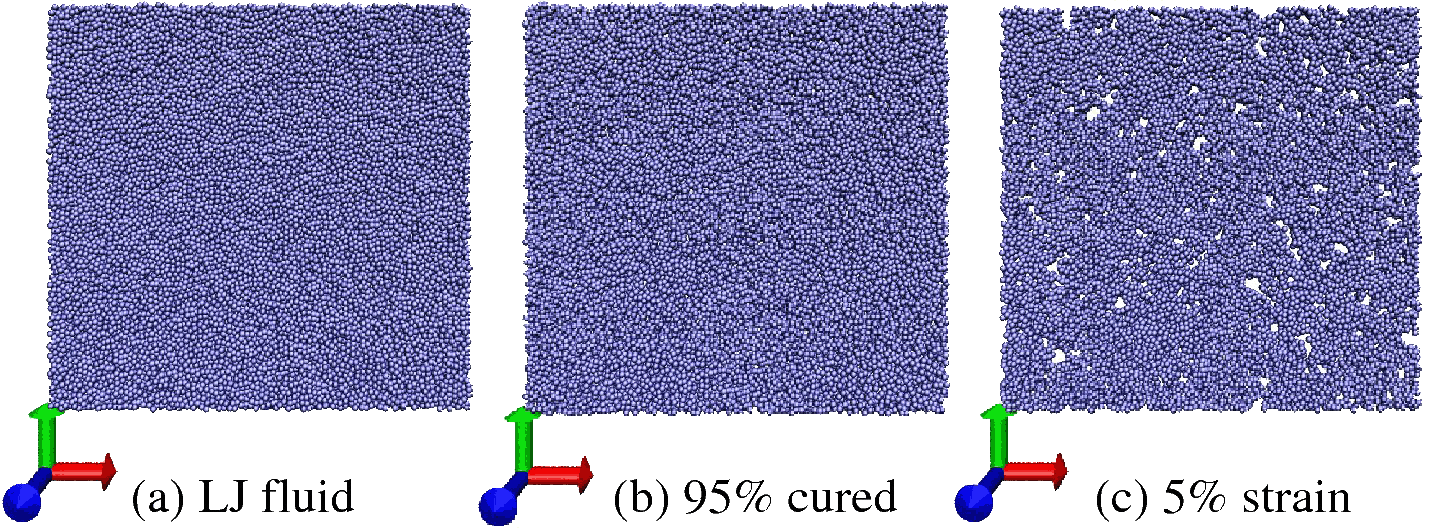
Highly cross-linked polymer (HCP) networks, including epoxies and vinyl ester-based
thermosets, are becoming increasingly important light-weight engineering materials,
finding uses as high performance adhesives and multi-functional composite matrices.
Despite the significant development of HCP's with improved performance, one particular
property of HCP that limits their usefulness is their lack of ductility. For example,
fully cured epoxies can be strong (i.e., about 2 GPa tensile strength) but brittle
(failing at about 1% strain). We have used large scale molecular dynamics simulations of
a bead-spring model to study glassy state neat HCP under tensile deformation.
Our results showed that a neat HCP exhibit strain hardening,
which was attributed to the formation and growth of micro-voids.
Strain hardening makes a HCP network ductile. While present understanding suggests
that the ductility can be improved by reducing bond density, which however also significantly
reduces the tensile strength, our results show that ductility can be tuned, at will,
by changing the bond angle flexibility. This also keeps tensile strength unaltered. Snapshot shows
cross-sectional view of 2 LJ radius thick layers of pre-cured, post-cured and deformed
HCP sample . It is clearly evident that there were no voids immediately after cure.
However, voids start to develop upon inducing tensile deformation.
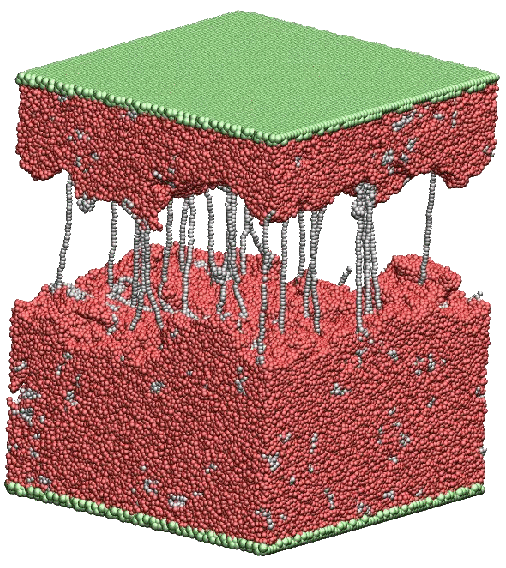 Mechanical properties of these thermosets can, however, be significantly modified by adding linear polymer chains, e.g., thermoplastics.
We have also studied thermoset/ thermoplastic polymer alloys tensile deformations. We focus here on the effect of the linear chain mass fractions,
linear chain lengths and strain rates on the mechanical properties of polymer alloys.
Our results show that tensile strain (i.e., strain to break) decreases with increasing
mass fraction up to a threshold value, beyond which it increases with mass fraction.
This non-monotonic behavior, which we call "anomalous ductility", is qualitatively
independent of strain rate and chain length. Threshold mass fraction decreases with
increasing chain length and we observe microscopic evidence that threshold value signifies
the onset of interchain interactions. A simple scaling argument suggests that threshold
mass fraction is related to the overlap concentration of the thermoplastic homopolymer
in the cured thermoset matrix. The snapshot is a space-fill representation of an alloy
of 8% mass fraction after fracture. In some cases we also shear the system and we find
a "slip-stick" type fracture. This behavior was attributed to the escape of
mono-dispersed homopolymer chains from the host cavities near the broken interfaces.
Mechanical properties of these thermosets can, however, be significantly modified by adding linear polymer chains, e.g., thermoplastics.
We have also studied thermoset/ thermoplastic polymer alloys tensile deformations. We focus here on the effect of the linear chain mass fractions,
linear chain lengths and strain rates on the mechanical properties of polymer alloys.
Our results show that tensile strain (i.e., strain to break) decreases with increasing
mass fraction up to a threshold value, beyond which it increases with mass fraction.
This non-monotonic behavior, which we call "anomalous ductility", is qualitatively
independent of strain rate and chain length. Threshold mass fraction decreases with
increasing chain length and we observe microscopic evidence that threshold value signifies
the onset of interchain interactions. A simple scaling argument suggests that threshold
mass fraction is related to the overlap concentration of the thermoplastic homopolymer
in the cured thermoset matrix. The snapshot is a space-fill representation of an alloy
of 8% mass fraction after fracture. In some cases we also shear the system and we find
a "slip-stick" type fracture. This behavior was attributed to the escape of
mono-dispersed homopolymer chains from the host cavities near the broken interfaces.
|
|