Research Interests
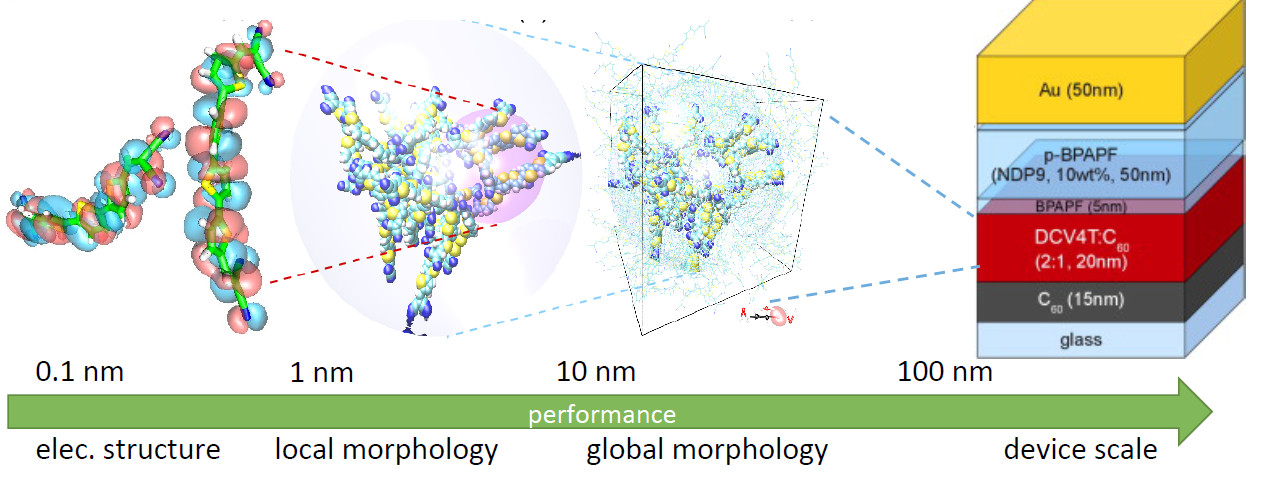
Technological advancements rely on the development of functional materials in a variety of conditions and compositions, ranging from perfectly ordered single-crystals to complex mixtures of soft matter systems. At the heart of their functionality lies, in general, the exploitation of effects occurring over a wide range of length- and time-scales, in and out-of equilibrium, from the nearly instantaneous excitation of a material after photon absorption to, e.g., extremely slow collective structural relaxations. Interest in the field of organic electronics, in particular, is largely provoked by the possibility to fine-tune properties of organic semiconductors by varying their chemical structure. Developing nanostructured materials for specific applications needs to account for the interplay of molecular electronic structure and material morphology, which requires the understanding of physical processes occurring on a microscopic level. The overall goal of designing cost- and conversion-efficient organic photovoltaic devices, for instance, faces several central challenges such as- optimization of charge photogeneration in donor-acceptor nanostructures,
- reduction of charge carrier recombination (geminate or non-geminate),
- optimization of charge carrier transport.
- the analysis of the interplay between molecular electronic structure and material morphology,
- an understanding of the microscopic quantities influencing charge and energy transfer,
- microscopic insight into light-energy conversion processes,
- interpretation of the charge and energy dynamics on a macroscopic (device-size) scale,
- structure-property relationships for optimizing device performance.
Microscopic simulations of charge and energy dynamics
The elementary focus of my research is the microscopic simulation of charge and energy dynamics in organic semiconductors. Such simulations combine first-principles electronic structure theory, molecular mechanics methods, charge transfer theory, and kinetic Monte-Carlo algorithms to obtain charge/energy dynamics in partially disordered organic semiconductors without the use of fitting parameters. The aim of these studies is to devise rules for the rational design/optimization of organic semiconductors with certain target properties, e.g., improve charge carrier mobility or reduce charge recombination. More specifically, this work includes method development for large-scale application [2], software design [3] (http://www.votca.org), and application to actual systems, e.g., to formulate design rules for charge transport efficient materials [4].The techniques developed herein are not restricted to the simulations in organic electronic materials, however. Charge and energy transport processes also play a significant role in biological macromolecular assemblies, e.g., in light-harvesting complexes for photosynthesis. Partitioning schemes, the coupling between quantum and classical regions, the calculation of transfer rates, and finally the dynamics can be applied to such macro systems. Similarly, an extension of the simulation framework to explicitly treat hybrid organic/inorganic nanostructures is a promising path of development for future projects.
Specifically, main current research directions and collaborations are summarized below:
Molecular excitations from Many Body Green’s Functions Theory (GW-BSE)
Method development currently focuses on the adaptation of the existing multiscale workflow for charge dynamics to the treatment of exciton processes in organic semiconductors. To this end, I concentrate on the evaluation and development of the underlying techniques based on many-body Green’s functions theory (GW-BSE) in collaboration with Prof. M. Rohlfing in Münster. An efficient localized-orbital based implementation of this beyond-DFT method coupled to standard packages of quantum chemistry allows to treat excited states of molecular structures containing >100 atoms with a high level of accuracy. Notably, it is possible to describe local and charge-transfer excitations, e.g. in donor-acceptor complexes, equally well with a single approach [5]. The current challenge is to account for environment effects when the complex is embedded in a molecular aggregate, e.g., formed by vapor-deposition or solution processing of the material. We found in a recent study [6] that mesoscale methods (polarizable continuum and/or lattice models) are inadequate to quantitatively account for local crystal fields and the related polarization induced effects. We therefore presently focus on the development of a hybrid QM/MM approach that couples the efficient GW-BSE calculations to a classical environment represented by atomic partial charges and polarizabilities.Charge photogeneration in organic photovoltaic cells
In a collaboration with the groups of Prof. K. Leo (IAPP Dresden) and Prof. K. Meerholz (U Köln) within the BMBF project MEDOS, we aim to use the above mentioned methodologies to understand the microscopic processes that affect the power conversion properties of organic solar cells based on acceptor-substituted oligothiophenes or merocyanine derivatives as donor material. We put particular emphasis on the analysis of the interface between donor and acceptor (fullerene) phases and the role interfacial excitations play in the generation of free charge carriers after optical excitation. Here, it is vital to support the experimental efforts by evaluating the conditions beneficial (detrimental) for charge generation and extraction (recombination) on an atomistic level [7]. This includes, on the one hand, the determination of the molecular structure of the interface and its dependence on processing conditions, such as substrate temperature during vapor-deposition, via atomistic molecular dynamics simulations, and the electronic structure of bulk and interface regions, on the other hand. Both, morphological order and concomitant electronic structure then influence rates for conversion between different states of the system and, therefore, the dynamics of charges and excitations in the actual device. Correlating morphology, electronic structure, and macroscopic device properties will eventually allow for a controlled development of new functional materials. A related project in the framework of the DFG Priority Program 1355 “Elementary Processes in Organic Photovoltaics” similarly deals with the photogeneration of charges in organic solar cells in which the donor phase is formed by a polymer material (PCPDTBT). Together with experimental collaborators at the MPI-P (Dr. F. Laquai), University of Potsdam (Prof. D. Neher) and University of Würzburg (Dr. C. Deibel), the intention is to use our computational approach to understand the origin of the differences in power conversion efficiencies of solar cells with silicon- or carbon-bridged derivatives of PCPDTBT. Specifically for the modeling of excitations, this is challenging due to the size of the polymer and the subsequent high demands for treating the quantum part. Dynamically varying conjugation lengths in the donor phase at room temperature and the effect of charge localization need to be accurately accounted for in both the quantum and classical parts of the simulation.Design of efficient host/guest systems for blue OLEDs
The available set of methods can also be used to study the electronic structure and related conditions for the desired recombination characteristics of charges in host/guest mixtures used in (deep-blue) light-emitting diodes. While the challenges for the simulation are largely identical, design objectives are in many respects inverted as compared to those for photovoltaics. Emissive recombination of charges is desired and the development focuses on the design of efficient and stable blue emission [1,4]. This research is performed together with industrial partners (Dr. C. Lennartz, BASF) within the BMBF project “Morphologie und elektronische Struktur von Organik/Organik und Organik/Metalloxid-Hybridsystemen” (MESOMERIE).Scale-bridging by stochastic modeling
Our microscopic simulations are limited by the accessible length- and time scales, which make a treatment of, e.g., a realistic morphology of a bulk heterojunction active layer of a solar cell impossible. As an alternative to systematic coarse-graining, I am working with the group of Prof. V. Schmidt (Ulm University) on stochastic models that allow for overcoming present limitations [10]. Additionally, we also collaborate with Prof. D. Kroese (University of Queensland, Brisbane) at improving Monte-Carlo algorithms for the rate-based dynamics.My current research profile heavily relies on the experience during my PhD in the field of surface and interface science in hard condensed matter systems. Here, I gained expertise in the first-principles modeling of structural and electronic properties of wide-gap semiconductors and insulators. In particular, my project dealt with approximate self-interaction corrections to density-functional theory with which it is possible to obtain accurate electronic band structures not only in bulk crystals, but also for structures in reduced dimensions, i.e., surfaces or nanotubes [9]. This experience is an essential basis for future projects that focus on coupled organic and inorganic materials, such as studies of electron injection from photoexcited dye-molecules into nanoparticles in dye-sensitized solar cells. During research projects at University of California Irvine, I have also accumulated experience in the field of plasmonics, in particular the scattering of surface-plasmon polaritons (SPP) from surface defects. Effects related to SPP will play an increasingly important role not only in the interpretation of scattering experiments. For instance, resonances of SPP can be exploited to increase the efficiency of solar cells. For this it is important to rationally design a desired target scattering behavior, along the lines of my previous work in which I demonstrated the possibility of designing a cloak from SPP by a circular arrangement of point scatterers [10].
| [1] | Baumeier, May, Lennartz, Andrienko, J. Mater. Chem. 22, 10971 (2012) | [2] | Baumeier, Kirkpatrick, Andrienko, Phys. Chem. Chem. Phys. 12, 11103 (2010) | [3] | Rühle, Lukyanov, May, Schrader, Vehoff, Kirkpatrick, Baumeier, Andrienko, JCTC 7, 3335 (2011) | [4] | May, Al-Helwi, Baumeier, Kowalsky, Fuchs, Lennartz, Andrienko, J. Am. Chem. Soc. 134, 13818 (2012) | [5] | Baumeier, Andrienko, Rohlfing, J. Chem. Theory Comput. 8, 2790 (2012) | [6] | May, Baumeier, Lennartz, Andrienko, Phys. Rev. Lett. 109, 136401 (2012) | [7] | Schrader, Fitzner, Hein, Elschner, Baumeier, Leo, Riede, Bäuerle, Andrienko, J. Am. Chem. Soc. 134, 6052 (2012) | [8] | Baumeier, Stenzel, Poelking, Andrienko, Schmidt, Phys. Rev. B 86, 184202 (2012) | [9] | Baumeier, Krüger, Pollmann, Phys. Rev. B 73, 195205 (2006); ibid. 76, 085207 (2007) | [10] | Baumeier, Leskova, Maradudin, Phys. Rev. Lett. 103, 246803 (2009) |