
Since solid state NMR spectroscopy is a very versatile method, a broad field of application can be covered. Recent projects of the solid state NMR group are given below:
- Local structure and molecular dynamics in thin films for molecular electronics.
- Structure and dynamics of self-organizing supramolecular conductors
- Site selective investigation of molecular dynamics via ²H MAS NMR
- Structure and dynamics of novel columnar functional materials
- Secondary structure and molecular dynamics of oligo peptides and polypeptide block copolymers
- Self-organization and molecular dynamics of donor materials for organic solar cells
- Influence of thermal history and processing on molecular dynamics and morphology in poly olefines
- Heterogeneous dynamic behavior of polymers in the melt state
- Local dynamics in a natural polymers: starch-water interactions
Structure and dynamics in thin films for molecular electronics.
Constantin Haese, Daniel Pinkal, Robert Graf, Paul Blom.
Organic semiconductors are a material class with high scientific interest due to their promising application possibilities, originating from their handy properties (light, flexible, transparent) together with the possibility of low-cost mass production.
In organic electronic thin film devices, the local morphology and phase separation of the molecules plays a crucial role for their opto-electronic properties. Typically, diffraction measurements (e.g. X-ray) are performed to reveal the packing behavior of such materials, but they can only probe longer range order. In contrast, solid state NMR is able to give insights into the local inter- and intra-molecular arrangements which can in principle give direct conclusions on possible local electronic processes even for amorphous materials.
Moreover, NMR approches have the potential to monitor changes in the local electronic structure upon sample preparation as well as degradation process in devices.
Structure and dynamics of self-organizing supramolecular conductors
Ümit Akbey, Robert Graf in cooperation with Prof. Virgil Percec, University of Pennsylvania, Philadelphia.
The preparation of conducting nanoscopic fibers based on the self-assembly of discotic functional units is an appealing aim as they may be incorporated in molecular electronic devices. In this category, perylene derivatives have attracted great attention due to their exceptional high carge carrier mobility. A detailed knowledge about their supramolecular structure and molecular dynamics is crucial for the understanding of their electronic properties.
¹H MAS NMR, combined with ¹H-¹H double quantum (DQ) NMR methods, and ¹H-¹³C correlation experiments are powerful methods to investigate the structural and dynamics properties of non-crystalline, partially disordered systems. These methods combined with fast-MAS provide the spectral resolution needed to assign specific resonances and to obtain site specific structural and dynamics information to elucidate supramolecular systems with pronounced π-stacking effects.
Distinct line-shapes have been observed in perylene derivatives currently under investigation, with variing spacer groups connecting the perylene core and the outer dendron. In the ¹H MAS spectra, additional resonances due to supramolecular organization of perylene molecules are observed. The presence of different packing arrangements in these systems with strong π-shifts, causes that the number of recorded resonances easily exceeds the number of chemically distinct sites. NICS (Nucleus Independent Chemical Shift) maps help to identify the packing arrangements or can be used to verify or refine X-Ray studies of these systems.
Site selective investigation of molecular dynamics via ²H MAS NMR
Christoph Deller, Robert Graf.
One way by means of which the structural and dynamic information inherent in the anisotropic interactions, e.g. dipolar and quadrupolar couplings, can be probed in a site-selective fashion, involves specific isotope labels, e.g. 2H, 13-C and 15-N. For a water soluble compound with acidic proton sites, however, deuterons can easily be introduced via chemical exchange with D2O. This enables selective investigations of more complex systems containing acidic proton sites.
As ²H is a spin-1 nucleus, the ²H resonance and lineshape is governed by the strong quadrupolar interaction providing detailed insight into molecular dynamics easily. Under MAS, the ²H spectrum consists of a series of spinning side bands (separated by rotation frequency) with intensities that reflect the envelope of the static quadrupolar lineshapes, while distinct deuteron sites are resolved based on their isotropic chemical shifts. Yet, the ²H quadrupolar couplings can be removed from fast MAS NMR spectra when the data acquisition occurs in a "rotor-synchronized" fashion.
Upon the onset of motion, the intensities as well as the linewidths will be affected by the quadrupolar couplings due to a motionally induced reorientation of the ²H quadrupolar tensor (typically in the range of 40 - 125 kHz) during one rotor period (typically 0.1 ms, corresponding to a MAS frequency of 10 kHz). At the end of each rotor period the molecular motion interferes with the formation of the rotational echo yielding broad lines.
We investigate rotational motions in model compounds at the appropriate temperature range. The numerical fit of the obtained spinning side band patterns then reveals the type as well as the timescale of the rotational "jumps". Our results reveal that for known jump geometries the linewidths encountered in the rotor-synchronized spectra are directly proportional to the corresponding jump rates, extracted from the analysis of the spinning sideband patterns. Thus, in conclusion, the measurement of rotor-synchronized spectra can be sufficient for the determination of activation energies.
This fact becomes particularly interesting, if selective deuteration renders more than one ²H-site accessible in more complex systems. Rotorsynchronized acquisition can than provide distinction of the sites via chemical shift resolution and account additionally for the determination of activation energies, particularly in hydrogen bonded systems.
References
K. Schmidt-Rohr, H. W. Spiess, Multidimensional Solid-State NMR and Polymers, Academic Press, London (1994)
S. E. Ashbrook, S. Wimperis, "Rotor-synchronized acquisition of quadrupolar satellite-transition NMR spectra: practical aspects and double-quantum filtration", Journal of Magnetic Resonance, 177 (2005) 44-55. (Download)
M. Cutajar, S. E. Ashbrook, S. Wimperis, "H-2 double-quantum MAS NMR spectroscopy as a probe of dynamics on the microsecond timescale in solids", Chem. Phys. Lett., 4-6 (2006) 276-281. (Download)
A. Hoffmann, I. Schnell, "Two-Dimensional Double-Quantum 2H NMR Spectroscopy in the Solid State under OMAS Conditions: Correlating 2H Chemical Shifts with Quasistatic Line Shapes", Chem. Phys. Chem., 5 (2004) 966-974. (Download)
Structure and dynamics of novell columnar functional materials
Mihail Mondeshki, Robert Graf in cooperation with Prof. Yves Geerts, Université Libre de Bruxelles, Belgium.
I. Structural and Morphological Study of a Phthalocyanine-Perylene Binary System
Solid state NMR spectroscopy in combination with DSC, optical microscopy, X-ray diffraction and AFM, was applied on mixtures of functionalized phthalocyanine (Pc) and perylene (PTCDI) derivatives to investigate the thermotropic, structural and morphological properties of the mixtures and its components. The significant core, as well as chains, mobility even in the crystalline state at ambient conditions, as determined by the REDOR based 1H-13C recoupling REPT-HDOR (recoupled polarization transfer heteronuclear dipolar order rotor encoding) technique, pointed out the lath-shaped perylene derivative being a plastic crystalline compound (core dynamics order parameter, S, presented in the form of the second Legendre polynomial, equal to 0.66). Slight heating to 35°C resulted in a remarkable change in the core S value (0.26), which on further heating remained constant. On the other hand at ambient conditions to 100°C no signal of the aromatic carbons of the disc-shaped liquid-crystalline phthalocyanine derivative could be detected, which phenomenon was attributed to destructive interferences of the frequency of the phthalocyanine core motion and the magic angle spinning frequency. However, at temperatures as high as 100-140°C all aromatic ¹³C sites, as well as some aliphatic ones were accessible for the NMR recoupling procedure. The measured phthalocyanine core order parameter was on average S=0.28. The addition of the ¼th perylene to the columnar LC phthalocyanine resulted in a dramatic improvement of the π-π stacking in the mixture determined independently by WAXS and 1H solid state NMR, as well as in an increase of approximately 50% of the phthalocyanine core dynamic order parameter (0.45), which extended even to the aliphatic OCH2 groups. Possible explanations for the improved π-stacking are the donor-acceptor interactions between the electron rich Pc and electron poor perylene, as well as the reduced spatial requirements of the alkyl chains in the mixture, being diluted on addition of the perylene.
II. Solid state NMR study of the supramolecular order and molecular dynamics of a homologous series of octafunctioanalized phthalocyanines
The columnar packing and molecular dynamics of two liquid-crystalline octaesters (a linear chain C8 and a branched chain C8,4) phthalocyanine (Pc) compounds, as well as the mother carboxylic acid derivative, were investigated by solid state NMR, WAXS, DSC and DFT numerical simulations. The H-bonds formed in the carboxyl derivative proved to be sensitive on the rotor frictional heating and increased inner pressure under the fast MAS, which lead to decomposition and rotor crashes. In contrast with earlier studies, the DSC trace of the linear C8 derivative showed a clear transition at 75°C with an enthalpy value of 51 J/mol. Temperature dependent ¹H-¹³C cross-polarization (CP) and ¹H-¹³C REDOR based recoupling NMR studies under fast MAS have been carried out and correlated with the phase transitions observed in the DSC trace to further investigate the nature of the two phases. The low temperature phase, which we describe as a crystalline phase, was characterized by a dynamics order parameter of the phthalocyanine core S~1, which dropped down to 0.45 after entering the mesophase. The Pc discs packed in columns as observed by the WAXS powder spectra and were on average rotated at an angle close to 45° with a slight lateral displacement, as determined by the ¹H-¹³C correlation NMR experiments in good accordance with the chemical shifts DFT simulations. The steric requirements and pronounced mobility of the branched chain of the C8,4 phthalocyanine derivative shifted the mesohase transition to significantly lower temperatures. Also, upon heating characteristic phase instability in the region of the inner core protons as well as the chain OCH2 moieties were observed in the ¹H spectra and attributed to the molecular dynamics of the compound.
References
G. Zucchi, B. Donnio, Y. H. Geertz, Chem. Mater. 17, p. 4273-4277 (2005).
Secondary structure and molecular dynamics of oligo peptides and polypeptide block copolymers
Mihail Mondeshki, Robert Graf in cooperation with Prof. G. Floudas, FORTH, Ioannina, Greece and Prof. K. Müllen, MPI-P.
The self-assembly mechanism and the associated molecular dynamics of a series of poly-L-lysine functionalized polyphenylene dendrimer melts are studied as a function of the core size (generation), functionality and polypeptide length using X-rays, solid state NMR, calorimetry and dielectric spectroscopy. A striking dependence of the polyphenylene self-assembly on the polylysine length is shown. In addition, the type (α-helix/β-sheet) of peptide secondary structure is controlled by the packing restrictions imposed by the polyphenylene core. We show that constrained poly-L-lysines can adopt different secondary structures from their linear analogues. The dynamic investigation revealed three dynamic processes: a glass transition, a slower process associated with the relaxation of α-helical segments and a glassy mode whose origin could be resolved by site-specific solid state NMR techniques. Solid state NMR studies further indicated a mobility gradient in going from the rigid peptide backbone to the side chains.
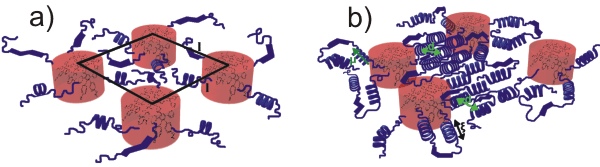
Schematic representation of the self-assembly in the functionalized polyphenylene dendrimers with (a) short polypeptides (N < 16) and (b) longer polypeptides (N > 20). Notice in the former the absence of a well-defined secondary peptide structure and that the polyphenylene dendrimers forming the core have some degree of order as revealed from SAXS. In the latter, the α-helical secondary structure prevails (with persistence length ξ), that in addition, display packing in a hexagonal lattice (with a distance d) (WAXS). Notice the absence of correlations between the cores in this case.
References
M. Mondeshki, G. Mihov, R. Graf, H.W. Spiess, K. Müllen, P. Papadopoulos, A. Gitsas, G. Floudas, "Self-assembly and molecular dynamics of peptide-functionalized polyphenylene dendrimers", Macromolecules 39, p. 9605-9613 (2006) (Download)
Self-organization and molecular dynamics of donor materials for organic solar cells
Martin Wegner, Robert Graf in cooperation with Prof. E.W. Meijer, Eindhoven University of Technology, Eindhoven, The Netherlands.
Supramolecular chemistry offers the possibility to create nanoscopic p/n heterojunctions by self-organization of electron donor and acceptor chromophores suitable e.g. for organic solar cells. At the present state of our studies, we focus the self-oranization and the molecular dynamics of various oligo(p-phenylene vinylene) (OPV) molecules. The influence of chiral and achiral aliphatic side chains as well as the influence of OPV chain length on their self-organization is studied using advanced solid state NMR methods under fast magic angle spinning.
Influence of thermal history and processing on molecular dynamics and morphology in poly olefines
Yefeng Yao, Shu Jie, Robert Graf, in cooperation with Prof. Sanjay Rastogi, Eindhoven University of Technology, Eindhoven, The Netherlands and Prof. Robert H. Gubbs, California Instisute of Technology, Pasadena, USA.
Material properties of polymers do not only depend on their chemical structure but also on the spatial organization of the molecules. For a simple polymer such as polyethylene, different organization of the chain molecules leads to a broad variety of macroscopic properties.
Advanced solid state NMR techniques recoupling chemical shift anisotropies and heteronuclear dipolar couplings are used to investigate the chain dynamics in chemically identical polyethylene samples with different morphologies obtained from different crystallization conditions or processing procedures. The experimental results demonstrate that the assumed direct relationship between local chain mobility in the non-crystalline region of sample and the cooperative chain motion between crystalline and non-crystalline regions is not fulfilled. Long scale cooperative chain motion, however, is crucial for the relaxation of local stress and thus determines mechanical properties of the material.
The Question, how the topology of apolymer chain affects local dynamics and morphology, is an open question. Therefore, high molecular weight PE samples with linear and ringshaped topoloy are compared.
Heterogeneous dynamic behavior of polymers in the melt state
Yefeng Yao, Robert Graf, in cooperation with Prof. Sanjay Rastogi, Eindhoven University of Technology, Eindhoven, The Netherlands.
The close spatial proximity of long polymers chains in the melt usually leads to topological constraints (entanglements) which determine the mechanical properties in the melt state. By controlled synthesis the number of entanglements in nascent material can be reduced. Ultimately, crystals composed of single chains are feasible, where the chains are fully separated from each other. If this separation can be maintained in the melt a new melt state can be formed.
The results from several NMR techniques combined with rheology studies show that through slow, carefully controlled melting such polymer crystals form a heterogeneous melt with more entangled regions, where the chains are mixed, and less entangled ones resulting from the melting of individually separated chains. Chain reptation, required for the homogenization of the entanglement distribution, is found to be considerably slow so that the heterogeneities in the melt are long-lived. The melt shows decreased significantly viscosity, and remarkably, provides enhanced drawability even in the semi-crystalline solid state. The procedure to obtain the heterogeneous melt state has been demonstrated from polyethylene. However, the protocol should be applicable to polymers in general.
Local dynamics in a natural polymer: starch-water interactions
Marianne Gaborieau, Robert Graf in cooperation with Prof. Michael J. Gidley, University of Queensland, Brisbane, Australia.
Starch is the main source of energy in human diet, as it is a major component of plants such as wheat, potatoes and rice. Furthermore, it has major applications in the paper, pharmaceutical and bipoplastics industries. Starch is a chemically simple polymer (it is a homopolymer of glucose), but it has a very complex macromolecular (branched) and supramacromolecular structure, with six identified hierarchical levels, on scales ranging from nm to mm. Starch granules contain water molecules, which is an integral part of the semi-crystalline structure. Starch-water interactions play a crucial role in the mechanical properties of starch-based bioplastics and also in the first stages of cooking of starch granules in excess water (gelatinization), therefore their understanding is of great interest to human and animal nutrition.
Partial sample deuteration by exchange of labile protons with deuterons provides an easy selective labeling of the sample. 2H solid-state NMR provides insight into starch-water interactions and will bring valuable information on the poorly understood endothermic process observed by DSC (differential scanning calorimetry) in many polysaccharides.