Tristan Bereau, Denis Andrienko , and O. Anatole von Lilienfeld, J. Chem. Theory Comput. (2015)
Parametrization of conformationally-dependent atomic multipoles without explicit QM calculation
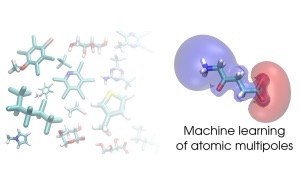
Accurate representation of the molecular electrostatic potential, which is often expanded in distributed multipole moments, is crucial for an efficient evaluation of intermolecular interactions. Here we introduce a machine learning model for multipole coefficients of atom types H, C, O, N, S, F, and Cl in any molecular conformation. The model is trained on quantum chemical results for atoms in varying chemical environments drawn from thousands of organic molecules. Multipoles in systems with neutral, cationic, and anionic molecular charge states are treated with individual models. The models' predictive accuracy and applicability are illustrated by evaluating intermolecular interaction energies of nearly 1,000 dimers and the cohesive energy of the benzene crystal.