Christian Kramer, Tristan Bereau, Alexander Spinn, Klaus R. Liedl, Peter Gedeck, and Markus Meuwly, J. Chem. Inf. Model. (2013)
Implementation of a multipole-fitting algorithm
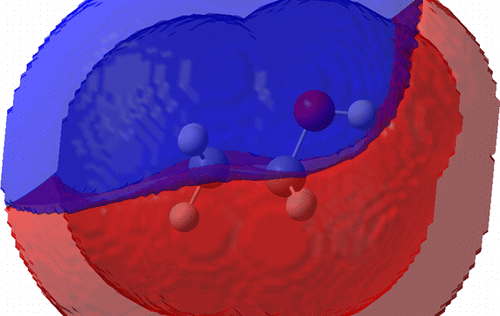
The description of molecular systems using multipolar electrostatics calls for automated methods to fit the necessary parameters. In this paper, we describe an open-source software package that allows fitting atomic multipoles (MTPs) from the ab initio electrostatic potential by adequate atom typing and judicious assignment of the local axis system. By enabling the simultaneous fit of several molecules and/or conformations, the package addresses issues of parameter transferability and lack of sampling for buried atoms. We illustrate the method by studying a series of small alcohol molecules, as well as various conformations of protonated butylamine.