Tristan Bereau, Robert A. DiStasio Jr., Alexandre Tkatchenko, and O. Anatole von Lilienfeld, Journal of Chemical Physics (2018)
Intermolecular interactions across chemistry from physics-based models and machine learning
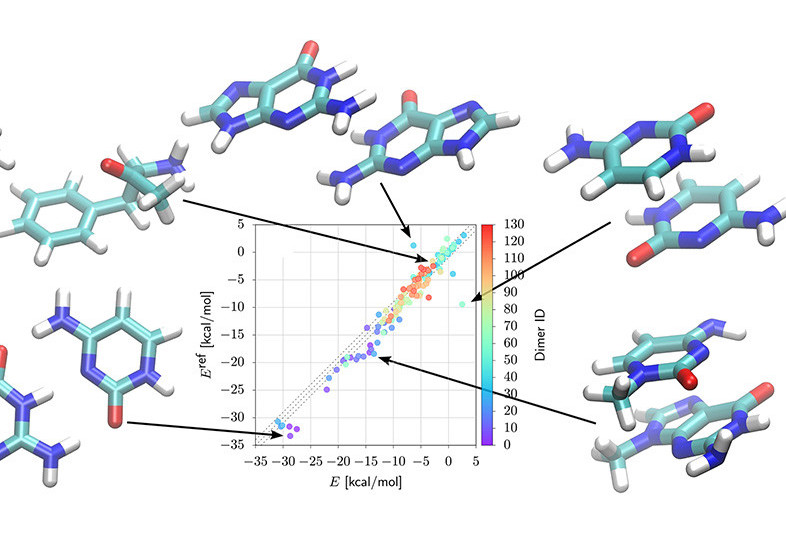
Classical intermolecular potentials typically require an extensive parametrization procedure for any new compound considered. To do away with prior parametrization, we propose a combination of physics-based potentials with machine learning (ML), coined IPML, which is transferable across small neutral organic and biologically relevant molecules. ML models provide on-the-fly predictions for environment-dependent local atomic properties: electrostatic multipole coefficients (significant error reduction compared to previously reported), the population and decay rate of valence atomic densities, and polarizabilities across conformations and chemical compositions of H, C, N, and O atoms. These parameters enable accurate calculations of intermolecular contributions—electrostatics, charge penetration, repulsion, induction/polarization, and many-body dispersion. Unlike other potentials, this model is transferable in its ability to handle new molecules and conformations without explicit prior parametrization: All local atomic properties are predicted from ML, leaving only eight global parameters—optimized once and for all across compounds. We validate IPML on various gas-phase dimers at and away from equilibrium separation, where we obtain mean absolute errors between 0.4 and 0.7 kcal/mol for several chemically and conformationally diverse datasets representative of non-covalent interactions in biologically relevant molecules. We further focus on hydrogen-bonded complexes—essential but challenging due to their directional nature—where datasets of DNA base pairs and amino acids yield an extremely encouraging 1.4 kcal/mol error. Finally, and as a first look, we consider IPML for denser systems: water clusters, supramolecular host-guest complexes, and the benzene crystal.
