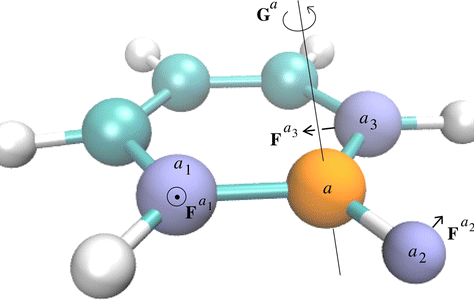
Tristan Bereau, Christian Kramer, and Markus Meuwly, J. Chem. Theory Comput. (2013)
Condensed-phase simulations with static atomic multipoles
Multipole (MTP) electrostatics provides the means to describe anisotropic interactions in a rigorous and systematic manner. A number of earlier molecular dynamics (MD) implementations have increasingly relied on the use of molecular symmetry to reduce the (possibly large) number of MTP interactions. Here, we present a CHARMM implementation of MTP electrostatics in terms of spherical harmonics. By relying on a systematic set of reference-axis systems tailored to various chemical environments, we obtain an implementation that is both efficient and scalable for (bio)molecular systems. We apply the method to a series of halogenated compounds to show (i) energy conservation; (ii) improvements in reproducing thermodynamic properties compared to standard point-charge (PC) models; (iii) performance of the code; and (iv) better stabilization of a brominated ligand in a target protein, compared to a PC force field. The implementation provides interesting perspectives toward a dual PC/MTP resolution, à la QM/MM.