Tristan Bereau and Kurt Kremer, J. Phys. Chem. B (2016)
Challenges in modeling the backbone in CG models
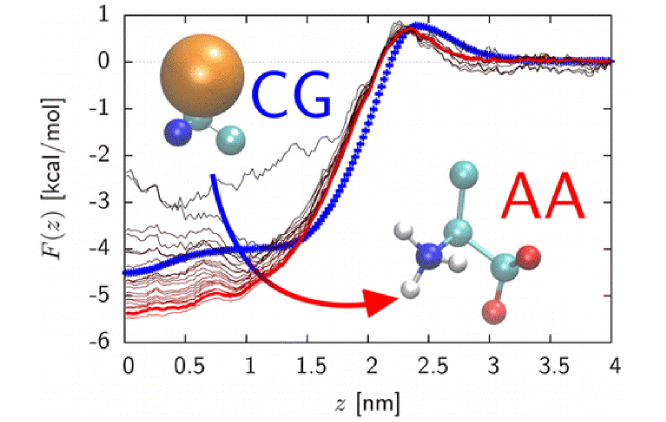
The thermodynamics of insertion of a protein in a membrane depends on the fine interplay between backbone and side-chain contributions interacting with the lipid environment. Using computer simulations, we probe how different descriptions of the backbone glycyl unit affect the thermodynamics of insertion of individual residues, dipeptides, and entire transmembrane helices. Due to the lack of reference data, we first introduce an efficient methodology to estimate atomistic potential of mean force (PMF) curves from a series of representative and uncorrelated coarse-grained (CG) snapshots. We find strong discrepancies between two CG models, Martini and PLUM, against reference atomistic PMFs and experiments. Atomistic simulations suggest a weak free energy of insertion between water and a POPC membrane for the glycyl unit, in overall agreement with experimental results despite severe assumptions in our calculations. We show that refining the backbone contribution in PLUM significantly improves the PMF of insertion of the WALP16 transmembrane peptide. An improper balance between the glycyl backbone and the attached side chain will lead to energetic artifacts, rationalizing Martini’s overstabilization of WALP’s adsorbed interfacial state. It illustrates difficulties associated with free-energy-based parametrizations of single-residue models, as the relevant free energy of partitioning used for force-field parametrization does not arise from the entire residue but rather the solvent-accessible chemical groups.