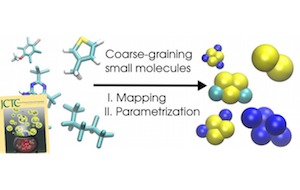
Tristan Bereau and Kurt Kremer, J. Chem. Theory Comput. (2015)
Quickly parametrizing the Martini force field of small organic molecules
The systematic exploration of chemical compound space holds many promises toward structure–function relationships and material design. In the context of computer simulations, progress is hampered by both the sheer number of compounds and the efforts associated with parametrizing a force field for every new molecule. A coarse-grained (CG) representation provides not only a reduced phase space but also a smaller number of compounds, due to the redundancy of CG representations mapping to the same structure. Though many CG models require the explicit force-field parametrization of a molecule with all others, others assume transferability by means of mixing rules, such as the Martini force field. To alleviate the burden associated with tedious parametrizations for each new compound, the present work aims at automating the mapping and parametrization of common small organic molecules for Martini. We test the method by analyzing the water/octanol partitioning of more than 650 neutral molecules, the hydration free energy of 354 others, and the free energies of hydration and solvation in octanol of another 69 compounds. Last, we compare with all-atom simulations the thermodynamics of insertion of four individual solute molecules in a phospholipid membrane. The protocol demonstrates the feasibility of an automated parametrization scheme for Martini and provides prospects for high-throughput simulation methodologies.
